Is the Complement System Irreducibly Complex?
by Mike Coon![]()
"By irreducible complexity I mean a single system which is composed of several interacting parts that contribute to the basic function, and where the removal of any one of the parts causes the system to effectively cease functioning. An irreducibly complex system cannot be produced gradually by slight, successive modifications of a precursor system, since any precursor to an irreducibly complex system is by definition nonfunctional."
-- Michael Behe
This essay is intended to back up some elements of Keith Robison's excellent FAQ detailing the shortcomings of Behe's arguments (http://www.talkorigins.org/faqs/behe/review.html). Another excellent rebuttal by H. Allen Orr can be found at http://bostonreview.net/br21.6/orr.html. See also http://bostonreview.net/evolution.html for several articles on intelligent design and irreducible complexity by Orr, Russell F. Doolittle, Douglas J. Futuyma, Richard Dawkins, Michael Behe, Phillip Johnson and David Berlinski among others.
Many people on Talk.Origins (T.O.) and elsewhere have outlined the conceptual reasons why Darwinian evolution can produce irreducibly complex (IC) systems. I try here to present some evidence to back up these concepts. In Robison's FAQ he'd mentioned that he did not know if the complement system showed an evolutionarily varying cascade system. Such variability across phylogenetic lines would illustrate the central fallacy behind Behe's concept of irreducible complexity. That fallacy stems from the inability to see that what now appears to be irreducibly complex is simply the result of Darwinian evolution whereby features that once were merely useful are now essential. Below I try to address Robison's point, and try to show how the complement system fails Behe's test.
By the way, one point that Robison made in his FAQ was in rebuttal to Behe's claim that antibodies cannot function on their own, and are therefore IC. Behe is wrong at least inasmuch as antibodies do have functions apart from the role they play in the complement cascade. There is even some evidence that antibodies have a function outside of their conventional "molecular tag" role. I will not go into details here, but a good for-instance would be the fact that antibodies can be catalytic. That is they can function not merely as molecular tags on pathogens, tumors or virally infected cells, but they can have enzymatic functions that are quite independent of their principle immunological roles (see, for example, Barbas1).
First, a brief review of the complement system2
In 1899 Belgian Jules Bordet discovered that serum contained two components, a heat stable one that had the activity of agglutination and precipitation, later identified by Paul Erhlich as antibodies, and a heat labile one that was responsible for bacterial lysis. However, it wasn't until the 1950s that the first system of heat labile serum proteins (more than 30 have since been identified) whose function served to complement the antibacterial action of antibodies was elucidated. Even earlier, though poorly understood, the role for the heat labile serum proteins was first noted in early attempts in blood transfusions and these observations can help us understand how complement works.
Most people know that one of the most important parts of our immune system is a kind of protein called an antibody. A specialized immune cell called a B cell makes antibodies. B cells are lymphocytes that play key roles in eradicating infections. Primarily, they develop into a kind of cell known as a Plasma Cell that produces large amounts of antibodies, although they also play a role as antigen presenting cells and in recruitment of T cells. Our antibodies are most frequently made to recognize invading pathogens and are thus not harmful to us. But sometimes antibodies can cause harm and clinicians refer to one way for this to happen as the hemolytic transfusion reaction, which is often fatal. The most common setting, rare these days, is transfusion of type A blood into a type O individual. The type O recipient makes antibodies against the red blood cells of the type A donor. Soon after transfusion, the recipient develops chills, fever, hives and uncontrolled bleeding. The recipient's plasma is pink, due to the leakage of hemoglobin from the donor red cells. The blood is depleted of clotting factors, because the clotting system has been activated and blood clots form diffusely within the circulation.
However, it turns out that binding of antibody to the type A red cells doesn't damage them. If we mix red cells suspended in saline with pure antibody, the cells clump, but they do not leak hemoglobin. When blood plasma is added to the clumped cells, the cells lyse, or burst open, and if we administer the plasma back to a recipient (in an animal model), it replicates the other features of a hemolytic transfusion reaction. Clearly, something in the plasma is carrying out these effects, not the antibody. This activity was termed "complement" because it complements the action of antibody. We now know a great deal about the makeup and the function of this important immune function and have identified three ways in which complement can kill invading pathogens.
Activated complement proteins initiate a cascade of enzymatic reactions that ultimately results in a large hole literally punctured through the membrane of the invading microorganism, or in the hemolytic transfusion reaction, in red blood cells. Our own cells are protected from spontaneously activated complement pathways (see below) because they possess other proteins that inactivate the complement cascade. Not surprisingly, some microorganisms have evolved a defense strategy that involves the presence of their own cascade inhibitors. But most do not have this defense and the protein complex that pierces the microorganism, called perforin, creates a large pore in the membrane of the pathogen. This loss of membrane integrity destroys the pathogen's ability to control the concentration of salts within itself, thus killing it.
Our complement effector functions serve as arms of both the adaptive and the innate immune systems. The innate immune system is found in all multicellular organisms while specific (or adaptive) immunity is found only in vertebrates, with the exception of the agnathans, or jawless fish. Antigen receptors in the innate system are germ-line encoded and recognize broadly homologous structures (usually essential) shared by groups of microbes. The adaptive immune system, however, develops its antigen receptor repertoire by somatic mutations and many of the determinants recognized by these antigen receptors are idiosyncratic (usually non-essential, and therefore mutable) to a particular microbe. The innate immune system, unlike specific immunity, does not develop memory. Both systems have the ability to distinguish self but the specific immune system is imperfect in this respect, resulting in autoimmunity.
The three complement pathways are weapons that an organism can use to help it fight off pathogens, destroy tumors and kill virally infected cells. They are known as the lectin, alternative and classical pathways (there are some minor variations in some organisms). The lectin, sometimes called the lytic, pathway and the alternative pathway are part of the innate immune response and are thus thought to be evolutionarily older than the classical pathway (see below). The lectin pathway is initiated by complement receptors comprised of circulating serum lectins (lectins are proteins that bind to sugar moieties), which bind pathogen surface molecules containing mannose residues (e.g. mannan), while the alternative pathway is initiated by complement that binds to the surface of a pathogen and is spontaneously activated. The third pathway, known as the classical pathway for historical reasons (it was discovered first), is activated by antibody opsonization of pathogens (binding of antibodies to specific epitopes of surface proteins, e.g. antigens) and is therefore a part of adaptive immunity.
Each of the three pathways has distinct early events including binding of receptors and a unique set of components of the complement system (see below) that initiate an enzymatic cascade. All three pathways converge at an enzyme activity called C3 convertase. For vertebrates, in both lectin and classical pathways the C3 convertase enzyme is a protein known as C2b while in the alternative pathway the convertase enzyme is a functionally and structurally homologous combination of two proteins, C3b and plasma factor B. This combination is in turn activated by plasma protease factor D. At this point all three pathways have similar effector functions. Below is a schematic of the major players in all three complement activation pathways of vertebrates* (please note: unfortunately, many of the proteins were numbered in the order they were discovered, not in their respective order in the pathways).
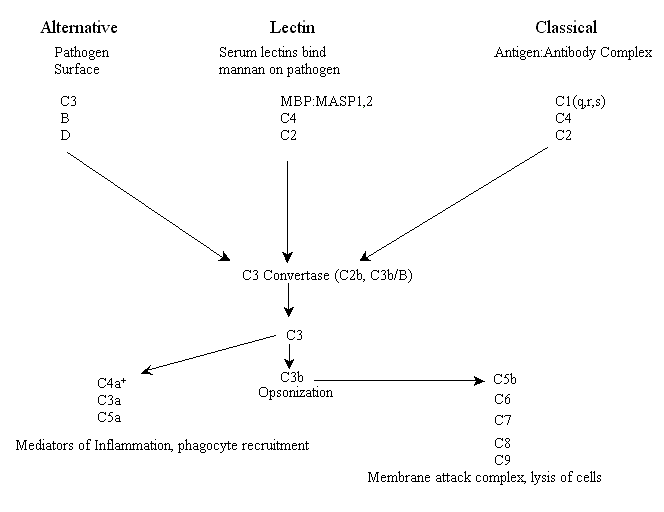
+C4a is a product of the cleavage of C4 by MASP or C1s, not C3, and it serves as a mediator of inflammation.
|
Summary of Proteins involved in the complement cascade |
|
|
Binding to Ag:Ab complexes |
C1q |
|
Activating Enzymes |
C1r, C1s, C2b, Bd, D, MASP1,2 |
|
Membrane-binding opsonins |
C4b, C3b, MBP |
|
Mediators of inflammation |
C5a, C3a, C4a |
|
Membrane attack |
C5b, C6, C7, C8, C9 |
|
Complement Receptors |
CR1, CR2, CR3, CR4, C1qR |
|
Complement-regulatory proteins |
C1INH, C4bp, CR1, MCP, DAF, H, I, P, CD59 |
*Adapted from Janeway & Travers Immunobiology, 1996; Current Biology Ltd/Garland Publishing Inc.
Evolutionary history of the complement pathways
All vertebrates that have been studied with the exception of the agnathans share the human complement systems. As we go further away phylogenetically, both the immune system and its effectors, including the complement systems, are different. Three that have been studied are the lamprey (an agnathan), the ascidian group of urochordates and the sea urchins. There has been a great deal of debate about the evolutionary history of the complement pathways. There is some evidence that in vertebrates the alternative pathway antedates the classical pathway, based upon sequence comparisons of the first component of complement, C1q3. However, other evidence suggests that the alternative pathway is older than the classical pathway based upon phylogenetic comparisons of C3a, C3b and C5a4. Moreover, the complement system of the lamprey consists only of the alternative and lectin pathways5. More recently, it has been shown that a urochordate has genes that correspond to the vertebrate C3, C4 & C5 genes, but not C26. Further, Ji et al7 and Nonaka, M et al8 showed that the lectin pathway is evolutionarily the oldest of the three by demonstrating that MASP is found in a urochordate, the ascidian Halocynthia roretzi (Japanese sea squirt). It is thought that much of the vertebrate complement system arose from duplication of genes encoding C3/C4/C5, Bf/C2, C1s/C1r/MASP-1/MASP-2, and C6/C7/C8/C9 molecules9
Is the complement pathway irreducibly complex?
One of the claims of Behe is that enzyme cascades like the activation of complement are irreducibly complex because each new step in the cascade "would require both a proenzyme and also an activating enzyme to switch on the proenzyme at the correct time and place". He points to the complement system, among others, as being irreducibly complex. And so it seems. For example, all three cascades rely upon the activity of C3 convertase in order to activate complement and help the organism fend off pathogens. Humans who lack C3 do not have functional complement pathways and suffer increased susceptibility to pyogenic infections from pathogenic bacteria like Haemophilus influenzae and Streptococcus pneumonae among others. It appears then that the vertebrate pathways could not have evolved in a Darwinian fashion from earlier lectin pathways, because they are dependent upon the activity of C3 convertase. But that's because Behe and others fail to realize that the current players in the cascade were not always present.
There are two general lines of evidence suggesting that the complement system evolved to its present status in mammals. One is that we see complement systems in invertebrates that lack a whole arm of the mammalian system. Another is that the proteins of the urochordate and vertebrate systems are homologous. In particular, there are homologies between proteases that suggest gene duplication and subsequent specialization, as noted above. The situation is similar to the clotting cascade, where homologies are observed that can be explained by gene duplication. Organisms that are phylogenetically far from humans, and thus represent a line of animals that are evolutionarily older than vertebrates today, nevertheless share the lectin pathway with us. This is one of a very great many pieces of evidence for common descent. Another is the fact that vertebrates possess an entire pathway that is absent in representatives of evolutionarily older organisms.
Let's look closer at the lectin pathway, the complement pathway shared by urochordates and vertebrates. It is initiated by the binding of mannan binding protein (MBP) and the MBP-associated serine protease (MASP), which is found as a proenzyme. MBP recognizes N-acetylglucosamine residues on surface proteins of pathogens, thus activating MASP. The activated MASP then proteolytically cleaves C4 and C2. C2 is cleaved to form C2b and C2a. C2b is also known as C3 convertase, which has enzymatic activity common to all three pathways. C3 convertase is critically important for mammalian complement pathways such that mutations in C2 (and C3) can produce disease states in rats, mice, guinea pigs, primates and humans (reviewed in 10-12). Deleting the C3 gene ablates the complement pathways as seen in knockout mice15.
The interesting thing about the sea squirt C3 protein is that it does not contain the C3 convertase cleavage site, found in all vertebrate C3 sequences to date7. This means that an enzyme that is dissimilar to vertebrate C3 convertase (C2b) activates the C3 protein of H. roretzi. In fact, urochordates do not have a C2 gene6. One candidate for the urochordate C3 convertase is MASP. MASP appears to be related by sequence homology to the C1 enzymes of the classical pathway one of which, C1s, cleaves C413, 14, thus suggesting a source for the vertebrate C1 genes through gene duplications. There are two MASP enzymes known to function as activating enzymes in complement pathways. MASP-1 binds to MBP (note: MBP is sometimes referred to as MBL for Mannan Binding Lectin). This binding activates MASP-1 which then cleaves C4 and C2, as noted above. It was recently postulated that MASP-1 also activates a second serine protease, called MASP-2, which produces a vertebrate C3 convertase enzyme in the lectin pathway13. A third MASP enzyme, MASP-3 has been identified and is thought to regulate the activity of the other MASPs15.
Can MASP itself activate C3 by functioning as a C3 convertase? Yes. Mammalian MASP can activate C3 directly, but it does so poorly16, 17 and mammals unable to convert C3 suffer from a variety of diseases10-12. Ascidian MASP, however, is known to have C3 convertase activity18. Since urochordates lack C2 and thus C2b, but do have MASP, they appear to have a truncated but obviously effective complement system bypassing the need for the C3 convertase C2b. So while vertebrate classical pathways are irreducibly complex in that the entire pathway is dependent upon at least one enzyme, it is clear that evolutionarily older organisms possess a functional pathway even in the absence of that enzyme. In the vertebrate lectin pathway, the C3 convertase C2b took over the function of the invertebrate MASP, such that now the vertebrate complement system is critically dependent on the activity of C2b.
This suggests that as the vertebrates arose and developed their own complement systems, they took advantage on the invertebrates' cascade, but added a component of their own, C2 (among others). This enabled the vertebrates to take full advantage of their new kind of immunity, adaptive immunity. They retained the lytic pathway, but also established the classical complement pathway by co-opting the invertebrate model. Over time, however, the vertebrate classical pathway became critically dependent on the new component, C2, which earlier pathways did not require. Vertebrates also developed the alternative pathway, once again co-opting the invertebrate pathway.
This bears repeating: urochordates have a functional complement system, yet they lack a component of the cascade, C3 convertase, which is essential in the same cascade in vertebrates. Recall Dr. Behe's quote at the beginning of this essay: "An irreducibly complex system cannot be produced gradually by slight, successive modifications of a precursor system, since any precursor to an irreducibly complex system is by definition nonfunctional". This example of an irreducibly complex complement cascade that is clearly the result of Darwinian evolution belies Dr. Behe's claim.
In summary, there are problems with Behe's claim that complement is an "irreducibly complex system" or that it is unlikely to have come about by evolution. Although it is highly articulated, the removal of some parts certainly doesn't make the system "cease to function". The occurrence of simpler systems in phylogenetically older organisms suggests an evolutionary pathway and the occurrence of numerous homologies between the genes suggests a mechanism for its evolution. This demonstrates the point that Keith Robison, H. Allen Orr, Russell Doolittle, Douglas Futuyma and a variety of T.O. regulars have tried to make over and over again. To paraphrase Dr. Orr (IIRC): "Darwinian evolution can easily produce irreducible complexity: all that's required is that parts that were once just favorable become, because of later changes, essential."19
Many thanks to George Acton for his knowledgeable suggestions and his critique of a draft of this essay.
References
3. Dodd, A.W., & F. Petry, Behring Inst Mitt 1993 (93):87-102.
4. Farries, TC, Steuer KL and JP Atkinson, Immunol Today 1990 (3):78-80.
6. Smith, L.C., Chang,, L., Britten, R.J., & E.H. Davidson, J., Immunol. 1996 (156):593-602.
7. Ji, X., Azumi, K., Sasaki, M.,& N. Masaru, Proc. Natl. Acad. Sci USA, 1997 (94):6340-6345.
9. Zarkadis IK, Mastellos D, Lambris JD, Dev Comp Immunol 2001 Oct-Dec;25(8-9):745-62
10. Sakamoto M, Fujisawa Y, Nishioka K Nutrition 1998 (4):391-8.
11. Carroll MC Annu Rev Immunol 1998 (16):545-68
12. Frank M.M., J Clin Immunol 1995 (6 Suppl):113S-121S.
13. Vorup-Jensen T, Jensenius JC, Thiel S. Immunobiology 1998 Aug;199(2):348-57
14. Matsushita M, Endo Y, Fujita T. Immunobiology 1998 Aug;199(2):340-7
16. Ogata, R.T., Low, P. J. & M. Kawakami, J. Immunol. 1995 (154):2351-2357
17. Matsushita, M. & T. Fujita Immunobiology 1995 (194):443-448
18. Nonaka M, Azumi K. Dev Comp Immunol 1999 Jun-Jul;23(4-5):421-7
19. See http://bostonreview.net/br21.6/orr.html.
[Return to the Irreducible Complexity FAQs]