Errores Plagiados y Genética Molecular
Otro argumento en la controversia entre evolución y creacionismo
por Edward E. Max, M.D., Ph.D.El siguiente ensayo es una actualización de un artículo que publiqué en Creation/Evolution en 1986 (XIX, p.34). Lo publico con permiso de Creation/Evolution.
![]()
|
Otros enlaces:
|
1. Introducción
La mayoría de los científicos considera que la evidencia a favor de la evolución es abrumadora. Por lo tanto, en su convicción de que la evolución ya ha sido documentada de manera exhaustiva y suficiente, a veces no consideran cómo los nuevos descubrimientos podrían proporcionar evidencia para la evolución que resulte poderosamente persuasiva para individuos que se inclinan hacia las creencias creacionistas. En este artículo, describo algunos descubrimientos de mi propio campo de la genética molecular, descubrimientos cuyas implicaciones para el debate entre creacionismo y evolución no se discutieron explícitamente cuando fueron reportados. Intento mostrar cómo proporcionan evidencia para la evolución que es tanto convincente como conceptualmente simple lo suficiente para que el público interesado la aprecie.
La nueva evidencia molecular se refiere a una cuestión que, en mi opinión, representa uno de los pocos casos en los que un argumento creacionista ha demostrado coherencia lógica y ha llevado la posición evolutiva a un punto muerto. Esta es la cuestión de cómo interpretar las similitudes entre las especies vivientes modernas, especialmente las similitudes observadas a nivel molecular. Como veremos, los recientes descubrimientos de la genética molecular resuelven este punto muerto de manera inequívoca a favor de la evolución.
1.1 La visión evolutiva de las similitudes entre especies
Consider first how evolutionists interpret similarities between species living today. Present-day humans and chimpanzees, despite obvious external and behavioral differences, have extremely similar internal organs and physiological functions; indeed their genes are more than 98% identical (Goodman et al., J Molec Evolution 30:260,1990). Just as the resemblance between two siblings suggests a common parentage, resemblance between species suggests common ancestors. Evolutionists believe that humans, gorillas, and chimpanzees evolved from a common ancestor: an ape-like creature that lived perhaps five to ten million years ago, rather recently on the geological time scale. (The thought that humans and apes might share a common ancestor seems particularly unacceptable to creationists because of the theological implications of such a relationship and the clear contradiction to the creationists' literal interpretation of biblical Genesis.) Species less similar to humans than are apes--mice, for example--are believed to have branched off millions of years earlier from a common primitive mammalian ancestor. Evolutionary family tree diagrams that express such relationships between species have been constructed by evolutionary biologists by analyzing similarities of present-day organisms. In many cases, fossilized remains of extinct species can be used to support the features of such evolutionary trees; fossil evidence will not, however, be discussed in this article.
Otra fuente extensa de datos que ha sido de gran importancia en la construcción de diagramas de árboles de similitud es la comparación de proteínas y genes entre especies. Las proteínas son grandes moléculas biológicas formadas por subunidades llamadas aminoácidos que se unen entre sí en cadenas, como los vagones de un tren. Existen veinte tipos diferentes de aminoácidos utilizados en las proteínas, y la mayoría de las proteínas contienen cientos de estas subunidades. Cada proteína tiene un número y secuencia específicos de aminoácidos, y esta secuencia determina las propiedades que tendrá dicha proteína. Para que una célula sintetice una proteína específica, debe acceder a un "banco de información" en el que se almacenan las secuencias de aminoácidos; este banco de información está compuesto por los genes del organismo, que contienen las secuencias de aminoácidos codificadas en moléculas de ácido desoxirribonucleico (ADN). Los bioquímicos pueden purificar proteínas y conocer la secuencia exacta de sus aminoácidos, o pueden obtener esta información leyendo la secuencia apropiada del ADN del organismo. Se ha dedicado un considerable esfuerzo a comparar las secuencias de proteínas similares aisladas de diferentes especies. Por ejemplo, una proteína llamada "citocromo c" ha sido examinada en más de ochenta especies. Estas secuencias de aminoácidos del citocromo c representan bits de datos "digitales" que pueden utilizarse para cuantificar las diferencias entre especies, y estas diferencias pueden emplearse para construir árboles evolutivos muy similares a los basados en comparaciones de características "analógicas" de la anatomía corporal. Tales árboles de secuencias de proteínas, así como los basados en similitudes de la estructura del ADN, concuerdan notablemente bien con los árboles evolutivos derivados anteriormente de similitudes anatómicas. La concordancia de los árboles evolutivos construidos a partir de tipos de datos completamente diferentes (por ejemplo, Goodman et al Mol Phylogenet Evol 9:585, 1998) ha sido tomada por los evolucionistas como evidencia de la validez del marco intelectual sobre el que se basan los árboles: la teoría de la evolución (véase Jukes, en Scientists Confront Creationism, editado por Godfrey, WW Norton, Nueva York 1983; Creation/Evolution XVIII:42, 1986; Goodman et al., J Mol Evol 30: 260, 1990).
1.2 La visión creacionista de las similitudes entre especies conduce a un callejón sin salida
Sin embargo, los creacionistas tienen una interpretación alternativa de las similitudes en las secuencias de aminoácidos reflejadas en los árboles de los evolucionistas. Dicen que tales similitudes en especies "relacionadas" simplemente reflejan la elección del creador de diseñar especies similares para funcionar de manera similar, no solo a nivel de huesos, músculos y órganos, sino también a nivel de la función de las proteínas; de ahí las similitudes en las secuencias de aminoácidos.
Por lo tanto, las similitudes entre especies en anatomía y estructura proteica pueden interpretarse de dos maneras completamente diferentes. Los evolucionistas dicen que la similitud entre características de, por ejemplo, humanos y simios refleja el hecho de que estas características fueron heredadas de un ancestro común; es decir, las características similares de humanos y simios están determinadas por copias modernas de genes que una vez existieron en una especie que fue ancestral tanto para simios como para humanos. Los creacionistas dicen que simios y humanos fueron creados independientemente pero fueron diseñados con características similares para que funcionaran de manera similar. Tanto la visión de copia de genes como la de creación independiente parecen consistentes con los datos de similitud, pero ¿cuál visión es correcta?
1.3 Una posible justificación para resolver el bloqueo
Una forma de distinguir entre copia y creación independiente se sugiere por analogía con los siguientes dos casos de la literatura legal. En 1941, el autor de un libro de texto de química interpuso una demanda alegando que partes de su libro de texto habían sido plagadas por el autor de un libro de texto competidor (Colonial Book Co, Inc. v. Amsco School Publications, Inc., 41 F. Supp.156 (S.D.N.Y. 1941), aff'd 142 F.2d 362 (2nd Cir. 1944)). En 1946, el editor de un directorio comercial para la industria de la construcción hizo cargos similares contra un editor de directorio competidor (Sub-Contractors Register, Inc. v McGovern's Contractors & Builders Manual, Inc. 69 F.Supp. 507, 509 (S.D.N.Y. 1946)). En ambos casos, la mera similitud entre el contenido de las copias alegadas y los originales no se consideró evidencia convincente de copia. Después de todo, ambos libros de texto de química describían el mismo cuerpo de conocimiento químico (los libros estaban diseñados para "funcionar de manera similar") y ambos directorios listaban miembros de la misma industria, por lo que se esperaría una similitud sustancial incluso si no hubiera ocurrido copia. Sin embargo, en ambos casos, los errores presentes en los "originales" aparecieron en las copias alegadas. Los tribunales juzgaron que era inconcebible que los mismos errores pudieran haber sido cometidos independientemente por cada demandante y demandado, y dictaminaron en ambos casos que había ocurrido copia. El principio de que los errores duplicados implican copia está ahora bien establecido en la ley de derechos de autor. (En reconocimiento de este hecho, los editores de directorios incluyen rutinariamente entradas falsas en sus directorios para atrapar a posibles plagiatas.)
¿Se pueden utilizar los "errores" en las especies modernas como evidencia de "copiado" de antepasados antiguos? De hecho, la respuesta a esta pregunta parece ser "sí", ya que recientes investigaciones de genética molecular han descubierto algunos ejemplos de los mismos "errores" presentes en el material genético de humanos y simios. Para comprender estos hallazgos es necesario saber un poco sobre el ADN, la molécula química en la que se almacena la información genética.
2.1 Fundamentos del ADN
En un aspecto, la estructura básica del ADN se asemeja a la de las proteínas: ambas están formadas por cadenas lineales de subunidades variables. Aparte de esta característica común, la estructura del ADN es bastante diferente a la de las proteínas. Las subunidades en el ADN se llaman nucleótidos o bases, y la secuencia de estos nucleótidos contiene la información genética que especifica la secuencia de aminoácidos en cada proteína producida por el organismo. Mientras que 20 aminoácidos diferentes componen las subunidades de las proteínas, solo hay cuatro bases nucleotídicas diferentes en el ADN, generalmente abreviadas como A, T, G y C. Según el "código genético" deducido por los científicos en la década de 1960, cada aminoácido está especificado por uno o más tríos de nucleótidos; por ejemplo, la secuencia GCG especifica el aminoácido alanina. Dado que existen 64 tríos diferentes (cada uno llamado codón) y solo 20 aminoácidos para especificar, algunos aminoácidos están representados por más de un trío (por ejemplo, ATA, ATC y ATT codifican todos para el aminoácido isoleucina); y tres tríos —TAA, TAG y TGA— representan "codones de parada" que marcan el final de la secuencia del gen que puede utilizarse para especificar la secuencia de aminoácidos.
|
|
|
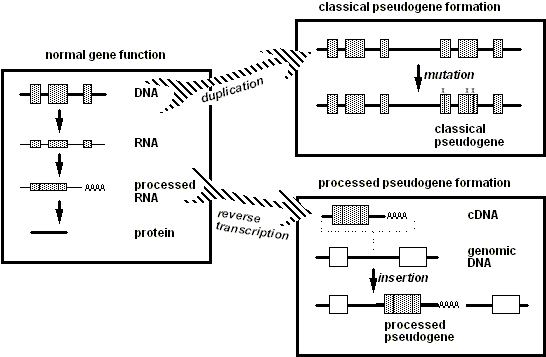
Figura 1. Fundamentos del ADN. El óvalo central representa una célula, dentro de la cual se encuentra el núcleo. Dentro del núcleo, la mayor parte del ADN existe como una doble hélice. El óvalo en la parte superior izquierda muestra una vista ampliada del ADN, en la que las hélices han sido dibujadas "desenrolladas" para revelar la similitud con una escalera. La información genética se almacena en las secuencias de bases nucleotídicas (A, T, G o C) que forman los peldaños de la escalera. Cada peldaño está formado por un par de bases nucleotídicas que se tocan entre sí, una base unida al esqueleto de una cadena y la otra al esqueleto de la otra cadena. Un nucleótido "A" siempre se empareja con un "T," y un "G" siempre se empareja con un "C." Para sintetizar una proteína, la célula lee la información genética del gen de esa proteína mediante la "transcripción" de una molécula de ARN desde el gen. Para la transcripción, las cadenas de la doble hélice del ADN deben separarse parcialmente para que las bases que forman el ARN puedan ensamblarse según las reglas de emparejamiento de bases complementarias. La vista ampliada en la parte superior derecha muestra las dos principales diferencias entre el ARN y el ADN: la cadena del esqueleto del ARN tiene una estructura química ligeramente diferente (representada por la línea punteada), y una forma ligeramente modificada de "T" conocida como "U" se encuentra en el ARN. La cadena transcrita de ARN actúa como un "mensajero" que transporta la información genética desde el almacenamiento en el núcleo hasta los módulos de fabricación de proteínas (representados en la figura por óvalos grises dobles) en el citoplasma. La vista ampliada en la parte inferior izquierda muestra que la secuencia de bases de ARN se lee de tal manera que cada tríada de bases especifica un aminoácido (aa1, aa2, etc.) en la proteína. La proteína se pliega en una estructura tridimensional funcional que depende de la secuencia lineal de aminoácidos. |

El ADN contiene dos cadenas lineales en una estructura de doble cadena que se asemeja a una escalera retorcida: la famosa "hélice doble". Los travesaños verticales de la escalera representan una cadena de esqueleto uniforme que no contiene información de secuencia. Como se muestra en la Figura 1 anterior, la información se almacena en los "travesaños" de la escalera, que están formados por un par de bases nucleotídicas, cada una sobresaliendo de una cadena vertical del esqueleto y tocando la base de la cadena opuesta para formar un "travesaño". La base G en una cadena siempre contacta con una base C en la cadena opuesta; de manera similar, una A siempre contacta con una T. Por lo tanto, una cadena de Ts en una cadena puede "emparejarse de bases" o "anclar" con una cadena que contiene una cadena de As para formar una estructura de doble cadena. La secuencia de bases nucleotídicas en una cadena se dice que es "complementaria" a la secuencia de la otra cadena. Para cualquier gen, los tripletes de bases que codifican la secuencia de aminoácidos se encuentran solo en una cadena. Algunos genes están codificados en una cadena, mientras que otros genes se encuentran en la otra cadena. En la mayoría de los genes de mamíferos, el ADN que codifica las secuencias de aminoácidos está interrumpido por segmentos de ADN aparentemente sin sentido ("intrones"). Las secuencias de intrones deben ser eliminadas antes de que la secuencia se utilice para ensamblar aminoácidos; esta eliminación, o empalme, no ocurre en la molécula de ADN, sino en la siguiente etapa de transferencia de información.
Para que una célula produzca una proteína particular cuya secuencia de aminoácidos está codificada en un gen, la información de la secuencia en el ADN debe primero copiarse o "transcribirse" en una molécula de cadena simple llamada ácido ribonucleico (ARN), como se muestra en la Figura 1 de arriba. Este transcripción inicial de ARN sufre varias alteraciones estructurales, conocidas colectivamente como "procesamiento", antes de ser utilizada para ensamblar aminoácidos. Estos pasos de procesamiento incluyen el "empalme" de segmentos intrón innecesarios del ARN y la adición de nucleótidos en un extremo--la "cola poli(A)"--que promueven el funcionamiento adecuado del ARN en la célula. Es el ARN "procesado" el que participa directamente en el ensamblaje de aminoácidos en proteínas. La transcripción de un gen en una copia de ARN está muy estrictamente controlada, en parte por secuencias regulatorias altamente específicas conocidas como promotores que, para la mayoría de los genes, ocurren en el ADN justo fuera de la región transcrita pero cerca de la posición donde debería comenzar la transcripción en ARN.
Cuando una célula se divide, toda la secuencia de su ADN debe duplicarse en dos copias fieles del original; una copia va a cada una de las células "hijas" creadas por la división. Ocasionalmente, ocurren errores en este mecanismo de copia, creando "mutaciones" en la secuencia de ADN. Existen varios tipos de mutaciones, incluyendo sustituciones de uno o pocos nucleótidos, deleciones de nucleótidos, duplicación de segmentos de ADN o inserción de segmentos de ADN extraños en una secuencia de ADN no relacionada. Tales cambios pueden ocurrir en la mayoría de las células del cuerpo—hígado, piel, músculo, etc.—sin ser transmitidos a la descendencia cuando el organismo se reproduce. Sin embargo, cuando las mutaciones ocurren en el óvulo o el espermatozoide o, más generalmente, en "células germinales" (es decir, el óvulo o el espermatozoide más sus precursores embriológicos), pueden ser transmitidas a las generaciones futuras. A menudo, las mutaciones son inconsecuentes: por ejemplo, pueden caer fuera de un gen, o si están dentro de un gen, pueden no cambiar el aminoácido codificado. Muchas diferencias genéticas entre especies estrechamente relacionadas se consideran tales mutaciones aleatorias e inconsecuentes. A veces, sin embargo, las mutaciones dañan críticamente la función de un gen. De hecho, tales mutaciones son la causa de enfermedades genéticas como la fibrosis quística, la anemia falciforme, la fenilcetonuria y cientos de otras, así como muchas aberraciones genéticas estudiadas en animales de laboratorio. Cuando los genetistas moleculares examinan el ADN de pacientes con tales enfermedades bien caracterizadas, casi siempre pueden encontrar el gen defectuoso e identificar la mutación que lo inactivó, ya que es raro que una enfermedad genética sea causada por una deleción que elimina un gen entero. Las mutaciones que causan enfermedades genéticas y malformaciones son generalmente tan perjudiciales para la supervivencia y el éxito reproductivo del organismo que en la naturaleza—es decir, en ausencia de la ciencia médica moderna—tienden a ser "eliminadas" por la presión de la selección natural. Rara vez, las mutaciones pueden ser beneficiosas para un organismo: estos casos raros forman la base para las adaptaciones evolutivas que mejoran la "aptitud" de un organismo a su entorno.
2.2 Errores del ADN
La tecnología de ADN recombinante ha permitido en los últimos años a los científicos determinar la secuencia de nucleótidos en segmentos de ADN de muchas especies, y se ha acumulado información equivalente a varios miles de millones de nucleótidos. Estas secuencias han aumentado enormemente nuestra comprensión de cómo funcionan normalmente los genes; pero, más al punto de este artículo, han proporcionado un tesoro de "errores" genéticos que son pistas potenciales para el análisis de la copia discutido anteriormente. En este contexto, uso la palabra "error" para incluir cualquier característica del ADN que tengamos buenas razones para creer que (1) originó de un "accidente" genético; (2) no sirve de beneficio al organismo que porta las características; y (3) por lo tanto no puede razonablemente interpretarse como haber sido "diseñada". Discutiré varias clases superpuestas de estos "errores", que argumentan a favor de la evolución de maneras ligeramente diferentes. Una clase incluye "genes pseudogenes", o copias no funcionales dañadas de genes. Discutiré tres clases de pseudogenes, la última de las cuales se superpone con otra categoría más grande de "errores" genéticos conocida como retrotransposones, que también será discutida. Véase la Figura 2.
|
|
|
Figura 2. Este diagrama de Venn ilustra las clases de "errores" discutidas en esta publicación, excepto que la clase de "pseudogenes unitarios" (que es realmente un subconjunto muy pequeño de "pseudogenes clásicos") no se muestra en el diagrama. Los pseudogenes procesados representan la intersección del conjunto de pseudogenes y el conjunto de retroposones. |

2.2.1 Pseudogenes
a. Pseudogenes unitarios.
Los conejillos de indias y los primates, incluidos los humanos, se enferman a menos que consuman ácido ascórbico en su dieta. Para los humanos y los conejillos de indias, el ácido ascórbico es por tanto una vitamina (vitamina C), mientras que la mayoría de las otras especies pueden sintetizar su propio ácido ascórbico y por tanto no requieren esta molécula en su dieta. La razón por la que los humanos y los conejillos de indias no pueden fabricar su propio ácido ascórbico es que carecen de un gen funcional que codifique la enzima proteica conocida como L-gulono-gamma-lactona oxidasa (GLO), la cual es necesaria para sintetizar ácido ascórbico. En la mayoría de los mamíferos están presentes genes funcionales de GLO, heredados -según la hipótesis evolutiva- de un gen funcional de GLO en un ancestro común de los mamíferos. Según esta visión, las copias del gen GLO en las líneas evolutivas de los humanos y los conejillos de indias fueron inactivadas por mutaciones. Presumiblemente esto ocurrió por separado en los ancestros de los conejillos de indias y los primates cuyas dietas naturales eran tan ricas en ácido ascórbico que la ausencia de actividad de la enzima GLO no era una desventaja -no ejercía presión selectiva contra el gen defectuoso.
Los genetistas moleculares que examinan secuencias de ADN desde una perspectiva evolutiva saben que las grandes deleciones génicas son raras, por lo que los científicos esperaban que las copias mutantes no funcionales del gen GLO --conocidas como "pseudogenes"-- aún estuvieran presentes en primates y conejos de Indias como reliquias del gen ancestral funcional. Por el contrario, los creacionistas creen que los humanos y los conejos de Indias fueron creados independientemente de todas las demás especies y debieron haber sido "diseñados" para funcionar sin GLO. Si esto fuera cierto, no se esperaría que estas dos especies portaran una copia defectuosa del gen GLO. De hecho, se han detectado pseudogenes GLO tanto en conejos de Indias como en humanos (Nishikimi et al. J Biol Chem 267: 21967, 1992; Nishikimi et al. J Biol Chem 269:13685, 1994), consistente con la visión evolutiva; presumiblemente, pseudogenes relacionados también existen en primates no humanos que requieren vitamina C en la dieta. Los tipos de mutaciones encontradas en los pseudogenes humanos y de conejos de Indias son típicos de las que se observan en enfermedades genéticas como las mencionadas anteriormente. En este ensayo llamo a las secuencias de ADN GLO humanas y de conejos de Indias "pseudogenes unitarios" para distinguirlas de otros dos tipos de pseudogenes que ocurren en una especie que también posee una copia funcional del mismo gen (véase a continuación). Los lectores deben notar que el término "pseudogene unitario" se utiliza aquí por conveniencia; no existe una nomenclatura estándar para describir este tipo raro de pseudogene.
Los pseudogenes unitarios son relativamente raros; cada uno es como un defecto genético que afecta a todos los individuos de una especie. Pero estos genes defectuosos no corresponden a enfermedades genéticas porque si causaran síntomas significativos u otros desventajas a sus dueños, los individuos con genes intactos habrían ganado la competencia por la supervivencia y la reproducción hace mucho tiempo, eliminando así el pseudogene de la existencia. Desde una perspectiva evolutiva, los pseudogenes unitarios representan reliquias genéticas de genes cuya función era importante en especies ancestrales pero se volvieron innecesarias en la especie moderna; es decir, son secuencias de ADN vestigiales. La presencia de reliquias de pseudogenes no funcionales se explica fácilmente con el modelo evolutivo: son una consecuencia natural de mutaciones que no son eliminadas por la selección natural porque la función del producto génico se ha vuelto innecesaria. De hecho, este modelo predice que se deberían encontrar muchos pseudogenes unitarios si los científicos examinan los genes que especifican estructuras vestigiales, por ejemplo, genes que codifican estructuras oculares en especies ciegas como las topos o los habitantes de cuevas. (Un ejemplo que confirma esta predicción fue recientemente descrito en topos marsupiales: un aparente pseudogene unitario relacionado con el gen de la proteína de unión a retinoides interreceptores [Springer et al., PNAS 94:13754, 1997]. Un ejemplo conceptualmente similar en el ADN humano lo proporcionan las secuencias de ADN de receptores odorantes: aproximadamente el 70% de estas secuencias son pseudogenes, reflejando el estado casi vestigial de nuestra percepción olfativa en comparación con la de otras especies [Rouquier et al., Nat Genet 18:243,1998; Rouquier, et al. Human Molec Genet &:1337,1998; Sharon et al., Genomics 61:24,1999; Rouquier et al., PNAS 97:2871,2000; Glusman et al, Genome Res 11:685, 2001].) En contraste, no se esperaría la existencia de tales pseudogenes si cada especie fuera creada independientemente por un diseñador inteligente (a menos que ese diseñador estuviera intencionalmente simulando la evolución). Se conocen varios otros pseudogenes unitarios en humanos, incluyendo secuencias homólogas [es decir, similares y pensadas como derivadas de un ancestro común] a los genes que codifican la urato oxidasa (Yeldandi et al, Gene 109:2821, 1994; Wu et al, J Mol Evol 34:78, 1992), la alfa-1,3-galactosiltransferasa (Galili y Swanson, PNAS 88:7401,1991) y la proteína de superficie RT6, una ADP-ribosiltransferasa unida por glicofosfatidilinositol (Haag et al, J Mol Biol 243:537,1994). Podrían descubrirse más a medida que el Proyecto del Genoma Humano avance en el conocimiento de nuestra secuencia de ADN.
(Otro grupo interesante de pseudogenes unitarios son las secuencias polimórficas que son genes en algunos individuos y pseudogenes en otros, con frecuencias diferentes de pseudogenes en diversas poblaciones. Ejemplos incluyen los genes humanos para el receptor de quimiocina CCR5, para la alfa2-fucosiltransferasa Se, para los citocromos p450 2C19 y p450 2D6, para la enzima tiopurina metiltransferasa y para la lipoproteína apo(a). En estos ejemplos, la ausencia de las proteínas funcionales correspondientes es generalmente inconsecuente, pero puede volverse clínicamente importante —ya sea beneficiosa o perjudicial— en ciertas circunstancias ambientales o en combinación con otros genes. En algunos casos, el mismo producto génico puede ser beneficioso en algunas circunstancias y perjudicial en otras, de modo que la selección conduce a una frecuencia génica intermedia.)
b. Pseudogenes duplicados clásicos
Una clase mucho más amplia de pseudogenes parece surgir de errores en un patrón de alteración génica que ha sido importante en la evolución de genes funcionales normales: el patrón de duplicación y diferenciación (Ohta, Genome 31:304,1989; Holland et al., Dev Suppl 36:125, 1994). Este patrón es evidente a partir de la observación frecuente (en ADN de una variedad de especies) de bloques de secuencias que aparentemente han sido duplicados de tal manera que aparecen de lado a lado dos o más repeticiones de secuencias similares, es decir, en tándem (véase caja 1).
Presumiblemente, inmediatamente después de la duplicación, cada copia del gen tenía una secuencia idéntica. (Véase la Figura 3.) Pero a medida que las secuencias de ADN se copian de generación en generación, las mutaciones pueden acumularse de manera independiente en las copias de la secuencia duplicada, con varias consecuencias posibles.
|
|
|
Figura 3. En la función génica normal (panel izquierdo), el ADN se transcribe en ARN, el cual luego es "procesado" mediante la eliminación de intrones (las secuencias no codificantes entre las cajas grises) y la adición de una cola poli(A). El ARN procesado maduro se traduce luego en una cadena de aminoácidos para formar una proteína. Los paneles derechos ilustran las dos vías que generan el pseudogén duplicado clásico (arriba) y el pseudogén procesado (abajo). En la vía superior, la duplicación del ADN genera dos copias del gen completo (caja superior derecha), pero las mutaciones en una copia (representadas por las "x") la convierten en un pseudogén. En la otra vía, un transcrito de ARN procesado de un gen puede convertirse en una copia de cDNA (caja inferior derecha) que se inserta de nuevo en el ADN de la célula en una posición aleatoria del genoma, usualmente—como se muestra aquí—en el ADN espaciador entre genes (cajas blancas en la Figura). |
i. Algunas mutaciones pueden no tener ningún efecto en el funcionamiento del gen.
| CAJA 1 |
|---|
|
Los creacionistas argumentan comúnmente que las características que observamos en el ADN de las especies modernas—presumiblemente incluidas las secuencias repetidas en tándem—fueron diseñadas específicamente por un creador inteligente; mientras que los científicos consideran que las secuencias de repeticiones en tándem son el resultado de duplicaciones accidentales del ADN. Un argumento a favor de la visión científica es que podemos ver ejemplos de tales accidentes genéticos ocurriendo hoy en el ADN humano (así como en el ADN de especies de laboratorio) sin aparente intervención divina. Debido al sesgo de detección—that is, solo encontramos cosas donde buscamos—los mejores ejemplos estudiados de duplicaciones en humanos son aquellas que causan enfermedades. Una forma en que las duplicaciones del ADN pueden causar enfermedades es si solo parte de un gen se duplica, de modo que la proteína resultante tendría algunos aminoácidos repetidos, alterando así la estructura y la función de la proteína (Heikkinen et al., Am J Hum Genet 60:48, 1997; Hu y Worton Hum Mutat 1:3, 1992). Cuando las duplicaciones del ADN ocurren en células somáticas (es decir, fuera de la línea celular que contribuye a los óvulos y espermatozoides), no pueden transmitirse a las generaciones futuras, pero pueden causar problemas al individuo afectado; por ejemplo, se han reportado casos de cáncer que contienen duplicaciones parciales de genes en las células cancerosas, mientras que la duplicación está ausente en los tejidos normales del cuerpo (Schichman et al., Cancer Research 54: 4277, 1994), indicando que la duplicación ocurrió durante la vida del paciente afectado. Las duplicaciones grandes que involucran genes enteros pueden crear problemas clínicos si las copias adicionales de un gen funcional completo pueden producir efectos dañinos; aunque esto es inusual, un ejemplo bien estudiado es la enfermedad neurológica enfermedad de Charcot-Marie-Tooth tipo 1A, en la cual una copia adicional del gen conocido como PMP-22 parece ser el culpable. Se han reportado numerosos casos de CMT1A donde la duplicación está presente en el paciente afectado pero no en ninguno de los padres del paciente, indicando que la duplicación debe haber ocurrido en las células germinales de uno de los padres o muy temprano en el desarrollo embrionario del paciente (Eur Neurol 34:135, 1994). Estos ejemplos hacen claro que la duplicación de genes no es solo una construcción hipotética—invocada para explicar repeticiones en tándem que fueron creadas por eventos inescrutables en el pasado remoto—sino que es un proceso bioquímico natural que puede observarse hoy en humanos (también puede estudiarse en especies de laboratorio tan diversas como bacterias y moscas de la fruta; Lupski et al. Am J Hum Genet 58:21, 1996). Los genomas de los vertebrados modernos pueden reflejar evidencia de dos eventos antiguos de duplicación a nivel del genoma que duplicaron todo el contenido génico (Sidow Curr Opin Genet & Devel 6:715,1996; Endo et al, Gene 205:19, 1997; Pebusque et al. Mol Biol Evol:1145, 1998); duplicaciones similares más recientes han sido deducidas en ciertas plantas, ranas e incluso ratas (Gallardo et al, Nature 401:341, 1999). |
ii. Otras mutaciones pueden dar lugar a una proteína que tenga una función ligeramente diferente a la del gen original. De hecho, tal diferenciación de genes duplicados para desarrollar nuevas funciones en una copia parece explicar una parte significativa de la expansión en la complejidad de los genes de los organismos superiores. Por ejemplo, se cree que el gen para una proteína primordial transportadora de oxígeno se duplicó, dando lugar a genes separados que codifican la mioglobina (la proteína transportadora de oxígeno del músculo) y la hemoglobina (la proteína transportadora de oxígeno de los glóbulos rojos). Luego, el gen de la hemoglobina se duplicó y las copias se diferenciaron en las formas conocidas como ![]() y
y ![]() . Más tarde, tanto el gen de la hemoglobina
. Más tarde, tanto el gen de la hemoglobina ![]() como el gen de la hemoglobina
como el gen de la hemoglobina ![]() se duplicaron varias veces, produciendo un grupo de secuencias relacionadas con la hemoglobina-
se duplicaron varias veces, produciendo un grupo de secuencias relacionadas con la hemoglobina-![]() y un grupo de secuencias relacionadas con la hemoglobina-
y un grupo de secuencias relacionadas con la hemoglobina-![]() . Los grupos incluyen genes funcionales que son ligeramente diferentes, que se expresan en momentos diferentes durante el desarrollo del embrión hasta el adulto, y que codifican proteínas específicamente adaptadas a esos períodos de desarrollo. La divergencia entre la mioglobina y los genes
. Los grupos incluyen genes funcionales que son ligeramente diferentes, que se expresan en momentos diferentes durante el desarrollo del embrión hasta el adulto, y que codifican proteínas específicamente adaptadas a esos períodos de desarrollo. La divergencia entre la mioglobina y los genes ![]() y
y ![]() ocurrió hace tanto tiempo en la evolución que la mezcla de información genética que ocasionalmente ocurre en el ADN ha distribuido estos genes a diferentes cromosomas. Los genes dentro del grupo
ocurrió hace tanto tiempo en la evolución que la mezcla de información genética que ocasionalmente ocurre en el ADN ha distribuido estos genes a diferentes cromosomas. Los genes dentro del grupo ![]() y el grupo
y el grupo ![]() se duplicaron más recientemente en la evolución y aún se encuentran en grupos.
se duplicaron más recientemente en la evolución y aún se encuentran en grupos.
iii. Finalmente, otras mutaciones que alteran aminoácidos críticos, que afectan el empalme de intrones o que crean nuevos codones de parada, pueden destruir completamente la función de una secuencia génica duplicada y convertirla en un pseudogén; de hecho, este es el destino de la mayoría de las copias génicas duplicadas (Lynch y Conery, Science 290:1151, 2000). Los tipos de mutaciones que destruyen la función génica nuevamente se asemejan a aquellas que han desactivado genes no duplicados cruciales, causando así enfermedades genéticas. Los genes defectuosos que no están duplicados tienden a desaparecer de las poblaciones con el tiempo porque los individuos que carecen de una copia funcional del gen son menos capaces de sobrevivir para producir descendencia (a menos que el gen ya no sea necesario, como en los pseudogenes unitarios). Sin embargo, cuando un gen defectuoso existe junto a una copia duplicada normal, la función continuada del gen normal generalmente compensa cualquier mutación en la copia defectuosa; la secuencia defectuosa suele ser inofensiva y puede perpetuarse en el ADN como un pseudogén "duplicado clásico". En general, cada pseudogén de este tipo contiene una secuencia que se asemeja al gen completo, incluyendo tanto las secuencias reguladoras que se encuentran fuera de las secuencias de codificación de aminoácidos como los intrones que interrumpen las secuencias de codificación (véase arriba). Se han encontrado numerosos pseudogenes de este tipo en el ADN de una variedad de organismos, incluidos los humanos. Por ejemplo, tanto los grupos alfa como beta de genes de hemoglobina en humanos incluyen pseudogenes duplicados de este tipo.
Aunque la mayoría de los pseudogenes "clásicos" se encuentran cerca del gen del cual originaron mediante duplicación en tándem, recientemente varios laboratorios han descrito una peculiar variedad de pseudogenes duplicados ubicados cerca de los centrómeros de varios cromosomas diferentes. (Los centrómeros son los segmentos cromosómicos donde, justo antes de la división celular, las dos copias duplicadas de los cromosomas en forma de salchicha parecen estar unidas, como se ilustra en la Figura 4, a continuación). Aparentemente, durante la evolución de los primates, varias regiones de ADN han sufrido un proceso poco comprendido que ha distribuido copias imperfectas a las regiones centrómeras de múltiples cromosomas. Los genes en estas copias incluyen intrones, pero en muchos casos están truncados y generalmente presentan múltiples mutaciones puntuales que los convierten en pseudogenes no funcionales. Ejemplos de estos pseudogenes centrómeros incluyen secuencias relacionadas con el gen de adrenoleucodistrofia ALD (Eichler et al., Human Molec Genet 6:991, 1997), el gen transportador de creatina SLC6A8 (Eichler et al., Human Molec Genet 5:899, 1996), el gen de neurofibromatosis NF1 (Human Molec Genet 6:9, 1997) y un gen llamado FRG1 cercano al locus de distrofia muscular facioscapulohumeral (Grewal et al., Gene 227:79, 1999).
c. Pseudogenes procesados
Una clase completamente diferente de pseudogenes conocida como pseudogenes procesados (véase la Figura 3, panel inferior derecho) surge de inserciones naturales de copias adicionales de genes derivadas de transcripciones de ARN. Tres características de estas secuencias sugieren un origen a partir de ARN:
i. Cada secuencia de pseudogén procesado se asemeja a un transcrito de ARN en que la similitud del pseudogén con su "gen fuente" se extiende desde el sitio de iniciación del ARN hasta el sitio de terminación del ARN, pero no incluye secuencias que se encuentran justo fuera de la región transcrita, incluidas las secuencias reguladoras como los promotores.
ii. Estos pseudogenes carecen de secuencias de intrones que normalmente se transcriben en ARN pero luego se eliminan del ARN antes de que se utilice para especificar la secuencia de aminoácidos de una proteína.
iii. Generalmente incluyen la cola "poly(A)" característica de los transcritos de ARN que codifican proteínas.
Además, a diferencia de los pseudogenes duplicados clásicos, que suelen encontrarse cerca del gen funcional del cual derivaron por duplicación, los pseudogenes procesados parecen insertarse en el ADN en ubicaciones aleatorias. Esta aleatoriedad es lo que se esperaría de una molécula de ARN que puede flotar libremente lejos de su gen fuente (del cual fue originalmente transcrito) antes de que una copia sea reinsertada de nuevo en el ADN. Incluso si codifica una secuencia correcta de aminoácidos, un pseudogen procesado suele ser no funcional porque carece de las secuencias reguladoras (como un promotor transcripcional) necesarias para la expresión génica; como una copia extra no funcional, tal secuencia puede acumular mutaciones aleatorias bajo ninguna presión selectiva, es decir, sin reducir el éxito reproductivo de un organismo que porta tales mutaciones.
Los pseudogenes procesados no deben confundirse con un pequeño número de copias de genes retrotrasladados que son realmente genes funcionales. Estos pueden surgir porque, raramente, una copia de ADN de un ARN procesado puede insertarse en el ADN de tal manera que la copia insertada pueda expresarse activamente. En estos casos, la secuencia de ADN puede retener su función y, por lo tanto, permanecer bajo presión selectiva contra la acumulación de mutaciones. Aunque tales secuencias —que pueden llamarse genes procesados o genes retrotrasladados— representan una fracción ínfima de las copias de genes retrotrasladados observadas, se han descubierto más de una docena, especialmente como copias de genes que se expresan específicamente en el testículo (Kleene et al, J Molec Evol 47: 275, 1998). Estos genes retrotrasladados se distinguen fácilmente de los pseudogenes procesados por su falta de mutaciones incapacitantes. El hecho de que estas pocas copias de ADN retrotrasladadas sean genes útiles no sugiere ninguna función para los mucho más numerosos pseudogenes procesados con múltiples mutaciones incapacitantes, como codones de parada, que impedirían su expresión como proteínas funcionales.
Los evolucionistas, tan pronto como Darwin, señalaron estructuras vestigiales—como los ojos sin función de animales ciegos que habitan en cuevas o los rudimentarios huesos pélvicos de algunas serpientes—como apoyo al punto de vista evolutivo. Estas estructuras no cumplen ninguna función aparente que pueda explicar su diseño por un creador, pero pueden entenderse fácilmente desde la perspectiva evolutiva como derivadas de estructuras que eran funcionales en especies ancestrales. Las secuencias genéticas vestigiales—that is, pseudogenes—proporcionan ejemplos exquisitos de estructuras vestigiales y, por tanto, evidencia especialmente convincente para la evolución. Tales secuencias pueden estudiarse en una variedad de especies; su relación con su contraparte funcional es obvia y cuantitativa (basada en el número de discrepancias de secuencia entre gen y pseudogen); y el subconjunto de pseudogenes procesados puede—con excepciones raras y fácilmente reconocibles—suponerse totalmente sin función desde el momento de su origen. Finalmente, los pseudogenes son una fuente rica de datos porque son abundantes. La secuenciación recientemente completada del cromosoma 22 humano (Dunham et al, Nature 402:489, 1999) identificó 134 pseudogenes junto con 545 genes en la región secuenciada, lo que corresponde a aproximadamente el 1.1% del genoma humano. Si esta muestra es representativa, podemos esperar aproximadamente 15000 pseudogenes en el genoma humano.
2.2.2 Retrotransposones
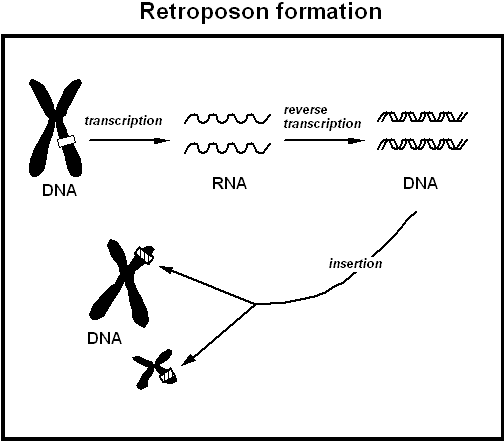
¿Cómo podría una secuencia de ARN procesado volver a incorporarse al ADN? De hecho, los pseudogenes procesados son miembros de una clase mucho más amplia de secuencias conocidas como elementos retrotraslocados, a los que aquí llamo "retrotransposones", y que todos implican secuencias de ARN que han sido insertadas en el ADN. Como se discutió anteriormente, en la fisiología molecular celular normal, la información genética pasa del ADN al ARN y luego a la proteína. Sin embargo, a veces ocurre un accidente genético y el ARN se transcribe al revés en ADN ("retro" o hacia atrás desde la dirección normal) y el ADN se deposita de nuevo (o "retrotraslocado") en alguna posición aleatoria del ADN de la célula. (Véase la Figura 4.) Algunos lectores pueden reconocer la retrotraslocación como el mecanismo mediante el cual retrovirus como el VIH--el virus del SIDA--se ocultan en el ADN de los linfocitos T de los pacientes con SIDA. Al igual que en el caso del VIH, un requisito crítico para toda retrotraslocación es la actividad de una enzima llamada transcriptasa inversa (RT), que genera una copia de ADN de la secuencia de ARN. No se conoce ningún gen que codifique esta enzima presente en el genoma humano, salvo en copias asociadas con elementos que sufren retrotraslocación. La enzima RT no tiene ninguna función conocida para la fisiología celular normal, aunque parece que una variante especializada de la transcriptasa inversa está involucrada en el mantenimiento de los telómeros, secuencias repetitivas en los extremos de los cromosomas. Una vez que una copia de ADN de un ARN ha sido sintetizada por RT, este ADN puede insertarse en las roturas del ADN que ocurren de vez en cuando en la célula y que normalmente son selladas por un complejo mecanismo de enzimas de reparación del ADN requerido por todas las células. Tales roturas a menudo ocurren en posiciones ligeramente diferentes en las dos hebras de ADN, produciendo "extremos escalonados"; las secuencias de retrotransposón insertadas entre tales extremos suelen estar flanqueadas en ambos lados por secuencias cortas idénticas creadas por la reparación de los dos extremos escalonados.
|
|
|
Figura 4. La formación de retroposones ocurre cuando una secuencia de ADN se transcribe en ARN, el cual luego es "transcrito al revés" de nuevo en ADN. En la parte superior izquierda, la Figura muestra un cromosoma tal como aparece justo antes de la división celular, pareciendo dos salchichas atadas juntas en el centrómero. Un gen en un cromosoma dado puede dar origen a varios pseudogenes, que generalmente se insertan aleatoriamente en diferentes cromosomas. Un mecanismo similar propaga los LINEs y SINEs, incluidas las inserciones Alu. |
De los muchos tipos de retrotransposones conocidos por los biólogos moleculares, mencionaré cuatro clases principales encontradas en el ADN humano. (Para una revisión reciente, véase Prak y Kazazian, Nature Reviews, 1:134, 2000.)
a. Pseudogenes procesados. En general, los pseudogenes procesados (descritos anteriormente) han sido descubiertos a medida que los científicos han examinado el genoma (utilizando técnicas fuera del alcance de este artículo) en busca de secuencias similares a genes conocidos. Tales cribas han revelado versiones mutadas con las características "procesadas" descritas anteriormente, lo que llevó a su identificación como retrotransposones. Como cualquier retrotransposón, esta clase solo podría haber entrado en el ADN de la línea germinal (es decir, el ADN de las células sexuales que permiten la propagación a futuras generaciones) si las células germinales contenían dos componentes: transcripciones de ARN del gen y transcriptasa inversa (RT) para copiarlo de nuevo a ADN. Consideremos estos dos componentes por separado. Muchas de las proteínas más conocidas se encuentran solo en tejidos específicos diferenciados y no se expresan en otros lugares. Por ejemplo, la hemoglobina se produce solo en células sanguíneas y sus precursores, y las proteínas de pigmento visual se producen solo en el ojo. Los genes de tales proteínas específicas de tejido casi nunca se transcriben en células germinales, por lo que raramente contribuyen a pseudogenes procesados. Por el contrario, todas las células tienen ciertas proteínas "de mantenimiento" necesarias para las funciones metabólicas básicas; las transcripciones de ARN que codifican estas proteínas están presentes en las células germinales y frecuentemente contribuyen a pseudogenes procesados. El gen de la gliceraldehído-3-fosfato deshidrogenasa, por ejemplo, es un gen de mantenimiento representado por aproximadamente 10 pseudogenes procesados en el ADN humano (Ercolani et al, JBC 263:15335, 1988). Como se mencionó anteriormente, las secuencias de pseudogenes procesados no incluyen las secuencias promotoras necesarias para la iniciación de la transcripción de ARN; por lo tanto, una vez que se insertan en el ADN, estos pseudogenes generalmente no se transcriben y, por consiguiente, no pueden representar por sí mismos un gen fuente para futuros retrotransposones. (Esta limitación contrasta con las características de otras clases de retrotransposones, como veremos a continuación.) El otro componente necesario para la retrotransposición es la transcriptasa inversa. La RT no está presente en la mayoría de los tejidos normales en cantidades medibles, aunque puede expresarse si una célula se infecta con un retrovirus que porta el gen para esta enzima. Las células germinales, sin embargo, son un tipo celular en el que se puede encontrar actividad de RT en ausencia de retrovirus infecciosos. En estas células, la enzima aparentemente deriva de otros elementos de retrotransposón —a discutirse a continuación— que portan genes funcionales de RT dentro de su secuencia.
b. SINEs. La clase mejor caracterizada de Elementos Dispersos Cortos (SINEs) en primates se conoce como secuencias Alu. Estas tienen aproximadamente 300 pb de longitud y no codifican ninguna secuencia de proteína. El reciente análisis de secuencias de ADN del genoma humano encontró aproximadamente 1,1 millones de Alus, que componen el 10,6% del ADN (Nature 409:860, 2001). A diferencia de los pseudogenes procesados, que generalmente no se transcriben, las secuencias Alu incluyen un segmento que puede actuar como un promotor transcripcional interno; por lo tanto, cada inserción Alu potencialmente puede transcribirse en ARN, sirviendo como fuente para una nueva inserción. Esta propiedad puede explicar parcialmente cómo estas secuencias se han vuelto tan abundantes en nuestros genomas. Sin embargo, la evidencia actual sugiere que solo muy pocas secuencias Alu son fuentes activas de transcritos; quizás la transcripción de la mayoría de las copias está inhibida por el entorno cromosómico de la inserción (Englander y Howard, JBC 270:10091, 1995). Incluso si los transcritos de ARN Alu existen en algunas células germinales, se reinsertan en el ADN solo raramente porque este paso requiere transcriptasa inversa, la cual puede no estar presente en las mismas células donde se está transcribiendo el ARN Alu.
c. LINEs. Los Elementos Dispersos Largos (Long Interspersed Elements) representan una familia de secuencias relacionadas que están presentes en aproximadamente 868.000 copias en nuestro ADN, constituyendo alrededor del 20% de nuestro genoma (Nature 409:860, 2001). Difieren de las secuencias Alu al ser mucho más largas—hasta aproximadamente 7000 pares de bases—y al contener dos secuencias codificantes potenciales. Una de estas secuencias codificantes presenta similitud con genes RT activos. Aunque en la mayoría de las copias de LINE el gen RT contiene numerosas mutaciones que impedirían que codifique cualquier enzima RT funcional, ciertas copias de LINE sí codifican transcriptasa inversa activa. Además, las regiones reguladoras justo fuera de las secuencias codificantes de los LINEs causan la expresión de los genes de manera selectiva en células germinales. Los LINEs tienen, por lo tanto, varias propiedades esperadas de las secuencias de ADN "egoístas" que pueden propagarse en el ADN del huésped simplemente porque codifican su propia maquinaria para propagarse. Los LINEs pueden expresarse en células germinales como ARN, y las copias raras que codifican RT funcional pueden permitir la transcripción inversa del ARN de LINE de nuevo a una copia de ADN, la cual puede luego insertarse en nuevas ubicaciones en el ADN de la célula germinales; cuando tal célula madura a óvulo o espermatozoide, puede ocurrir la transmisión del nuevo LINE a futuras generaciones. Aparentemente, la RT a menudo se desprende del ARN antes de que la transcripción inversa esté completa, ya que la mayoría de las copias de LINE están truncadas en sus extremos 5'. Es posible que la actividad de transcriptasa inversa codificada por LINE también pueda producir las copias transcritas inversamente de otros ARN—como transcripciones Alu y transcripciones de ARN de genes—que conducen a nuevas inserciones de secuencias Alu y pseudogenes procesados en el ADN celular.
d. Retrovirus endógenos. Los retrovirus infecciosos fueron descubiertos como agentes de enfermedades humanas y han sido estudiados intensivamente. Son los elementos retroposadores más complejos y pueden haber evolucionado a partir de otros más simples descritos anteriormente. Todos contienen dos Repeticiones Terminales Largas (LTRs) idénticas no codificantes en sus extremos, así como tres genes conocidos como gag, pol y env. Estos genes están codificados en el virus no por ADN sino por ARN. El gen pol codifica la transcriptasa inversa y también puede codificar actividades enzimáticas adicionales. El gen env codifica proteínas que recubren la superficie exterior (envoltura) del virus infeccioso. El gen gag codifica proteínas adicionales necesarias para procesar los componentes virales. La estructura común a todos los retrovirus es, por tanto, LTR-gag-pol-env-LTR. La LTR "izquierda" incluye secuencias reguladoras que pueden iniciar la transcripción de ARN hacia la derecha, hacia el gag-pol-env-LTR; la LTR "izquierda" es entonces recopiada de la LTR "derecha" por un mecanismo complejo. Los retrovirus infecciosos incluyen el HTLV-I, que causa un tipo de leucemia en humanos, y el VIH, que causa el SIDA. Estos virus típicamente infectan tipos específicos de glóbulos blancos—linfocitos—e insertan copias retrotranscritas de sus genes de ARN en el ADN de estas células. Poco después del descubrimiento de los retrovirus infecciosos, los científicos notaron que secuencias similares estaban presentes en el ADN de muchas especies mamíferas, incluidos los humanos; estas copias se llaman retrovirus endógenos y presumiblemente representan las consecuencias de antiguas infecciones retrovirales de células germinales. En el ADN humano hay aproximadamente 8 clases diferentes de retrovirus endógenos, con miembros de cada clase variando en número desde uno o dos hasta más de 50 copias. Esencialmente todos estos retrovirus endógenos contienen mutaciones que interrumpirían la función de sus genes, como cabría esperar si se insertaron hace millones de años sin presión selectiva para mantener la función de los genes. Además, las secuencias duplicadas de LTR representan objetivos potenciales para eventos de "recombinación homóloga" que eliminan el ADN entre la región correspondiente de las LTRs, dejando solo una secuencia compuesta de LTR; existen muchas más copias de estos fragmentos de LTR aislados en el ADN que copias de retrovirus completas.
3. Cómo pueden persistir errores antiguos en especies modernas
¿Cómo podrían cada una de las varias clases de secuencias no funcionales mencionadas anteriormente, que surgen en una célula germinativa de un individuo particular, llegar a ser preservadas en todos los individuos de una especie? Una posibilidad es que cada una de estas secuencias ocurrió por casualidad cerca de un gen ventajoso que se volvió prevalente en una población mediante la selección natural (el pseudogen o retroposón "montó a caballo" del gen ventajoso cercano; véase Nurminsky et al, Nature 396:572, 1998; Nurminsky et al. Science 291:128, 2001). Posiblemente tales secuencias no funcionales surgen a una frecuencia alta y solo vemos aquellas pocas que son preservadas por tales influencias indirectas o por eventos fortuitos en poblaciones pequeñas.
La carga adicional de transportar incluso una gran secuencia de pseudogen--por ejemplo, 100.000 nucleótidos--es insignificante para una célula mamífera con aproximadamente tres mil millones de nucleótidos de información. En cualquier caso, no existe ningún mecanismo conocido de "corrección de pruebas" mediante el cual la célula pueda distinguir el ADN no funcional del funcional y eliminar selectivamente lo que no necesita. Las secuencias de ADN sin función que los científicos han insertado en el ADN de ratones u otras especies se transmiten fielmente a la descendencia, y los pseudogenes y retrotransposones que ocurren naturalmente parecen comportarse de manera similar. La acumulación de ADN sin función no está completamente sin oposición; sí ocurren deleciones de ADN, pero aparentemente como accidentes raros que no discriminan entre secuencias funcionales y no funcionales. Las deleciones que eliminan genes funcionales cruciales han sido reconocidas como causas raras de enfermedades genéticas; la menor aptitud de los individuos que las portarían tendería a eliminar las copias de ADN con tales deleciones. Otras deleciones que por casualidad no eliminan ningún gen funcional podrían eliminar algo de ADN inútil, incluidos pseudogenes y retrotransposones; pero un individuo con tal deleción no tendría ninguna ventaja selectiva particular como resultado de la deleción, por lo que la propagación de copias de ADN que portan la deleción en la población en general no sería más probable que la propagación de cualquier otra mutación inconsecuente. Por lo tanto, tales eventos de deleción son claramente un mecanismo ineficiente de "eliminación de basura"; y, como consecuencia inevitable de esta ineficiencia, se han acumulado cantidades sustanciales de secuencias de "basura" sin función entre los genes funcionales de los mamíferos. Esta es una característica del material genético que no se apreció hasta que la tecnología de ADN recombinante permitió a los biólogos moleculares mirar más allá de las secuencias de aminoácidos hacia la estructura del ADN en sí. Aunque el alto contenido de "ADN basura" fue inicialmente sorprendente cuando se descubrió, nuestra comprensión actual de los mecanismos de expansión del genoma (duplicación e inserción) y la aparente falta de presión selectiva significativa para minimizar el tamaño del genoma se combinan para hacer que la acumulación de secuencias inútiles en nuestro ADN parezca inevitable.
4. El argumento del ADN a la evolución: pseudogenes compartidos y retrotransposones
La observación crucial que relaciona el descubrimiento de pseudogenes y retroposones con la teoría de la evolución es esta: algunos pseudogenes y retroposones se comparten entre diferentes especies, como si hubieran sido copiados de un pseudogene o retroposón en un ancestro común. Examinemos ejemplos de cada una de las clases de "errores" que hemos discutido anteriormente.
| CUADRO 2 |
|---|
|
Los pseudogenes de galactosiltransferasa compartidos son fascinantes por una razón que complica su uso para argumentar en contra de los creacionistas: la evidencia sugiere que hubo una ventaja selectiva para las mutaciones que inactivaron este gen. El producto enzimático del gen cataliza la producción de una molécula de carbohidrato específica que se encuentra en las membranas celulares de mamíferos que poseen la enzima, pero también en ciertas bacterias infecciosas. Los individuos infectados con tales bacterias se beneficiarían de montar un ataque inmunitario contra esta molécula de carbohidrato, pero si el mismo carbohidrato apareciera en sus propias células, tal ataque podría dañar sus propios tejidos. Por lo tanto, los individuos que portan mutaciones en la enzima--y por lo tanto no producirían el carbohidrato en sus propias células--estarían libres de montar un ataque inmunitario enfocado en esta molécula, protegiéndolos contra muchas bacterias sin peligro de dañar sus propios tejidos. Por lo tanto, la presión selectiva habría llevado a la diseminación de copias del gen que habían sufrido mutaciones incapacitantes. Los creacionistas podrían razonablemente argumentar que tales mutaciones podrían haber ocurrido independientemente en diferentes especies como ejemplos de microevolución reciente después de la creación independiente de las especies. Es posible que diferentes mutaciones hayan inactivado el gen independientemente en varios ancestros de primates. Sin embargo, los pseudogenes de galactosiltransferasa humana y chimpancé tienen mutaciones incapacitantes idénticas; por lo tanto, es más probable que el gen haya sido inactivado en un ancestro común humano/chimpancé. |
4.1. Pseudogenes unitarios compartidos. Muchos de los pseudogenes unitarios en humanos descritos anteriormente son compartidos con otros primates. Con "compartido" me refiero a más que simplemente que el mismo gen está inactivo en dos especies diferentes, ya que esa situación podría resultar si los genes correspondientes de las dos especies fueran inactivados por mutaciones independientes. En cambio, en todos los ejemplos que describo, los pseudogenes en primates portan muchas de las mismas mutaciones incapacitantes encontradas en los pseudogenes humanos correspondientes. Dado que mutaciones aleatorias independientes no serían probables que fueran idénticas en dos especies diferentes, los pseudogenes idénticamente mutados son una fuerte evidencia de que las mutaciones ocurrieron en una especie ancestral común.
Para el ejemplo del pseudogén unitario GLO de los humanos, se sabe que la vitamina C es necesaria en la dieta de otros primates, (aunque no para otros mamíferos excepto los cerdos de Guinea). La teoría de la evolución haría la fuerte predicción de que también se debería encontrar que los primates tienen pseudogenes GLO y que estos llevarían mutaciones incapacitantes similares a las encontradas en el pseudogén humano. Esta predicción fue enunciada en versiones anteriores del presente ensayo. Una prueba de esta predicción ha sido recientemente reportada. Una pequeña sección de la secuencia del pseudogén GLO fue recientemente comparada entre humano, chimpancé, macaco y orangután; se encontró que los cuatro pseudogenes comparten una delección de un solo nucleótido incapacitante común que causaría que el resto de la proteína se traduzca en el marco de lectura de tripletes incorrecto (Ohta y Nishikimi BBA 1472:408, 1999).
El gen RT6 mencionado anteriormente (2.2.1.a) codifica una proteína de aproximadamente 230 aminoácidos expresada en la membrana superficial de los linfocitos T de roedores; tanto el pseudogen humano como su homólogo chimpancé contienen mutaciones que producen los mismos tres codones de parada que impedirían la síntesis de una proteína RT6 (Haag et al, M Mol Biol 243:537,1994). Varios de los pseudogenes de receptores odorantes humanos mencionados anteriormente se encuentran en otros primates y comparten las mismas deficiencias que los pseudogenes humanos (Rouquier S et al., Nat Genet18:243,1998; Rouquier S, et al. Human Molec Genet &:1337,1998; Sharon et al., Genomics 61:24,1999). El pseudogen del receptor NPY1 humano comparte una mutación de marco crítico con los homólogos primates (Matsumoto et al., J Biol Chem 271:27217, 1996). El pseudogen de la urato oxidasa humana comparte tres mutaciones debilitantes con los pseudogenes chimpancé y orangután (Wu et al, J Mol Evol 34:78, 1992). Además, el pseudogen de galactosiltransferasa presente en el genoma humano se comparte con los simios y los monos del Viejo Mundo (Galili y Swanson, PNAS 88:7401, 1991), aunque la interpretación evolutiva de estos pseudogenes de galactosiltransferasa compartidos es compleja porque puede haber habido presión selectiva para inactivar esta enzima (véase Caja 2).
En resumen, aunque los pseudogenes unitarios son relativamente raros en los humanos, la mayoría de los ejemplos reportados se comparten con otros primates no humanos.
(Los únicos otros ejemplos de pseudogenes unitarios humanos que conozco son exclusivos de los humanos, habiendo adquirido aparentemente sus defectos incapacitantes después de la separación entre humanos y chimpancés; por lo tanto, son de interés como potenciales contribuyentes a las diferencias fisiológicas entre estas dos especies. Estos pseudogenes corresponden a una queratina del tipo I del cabello [Hum Genet 108:37, 2001], hidroxilasa de CMP-ácido siálico [Chou et al., PNAS 95:11751, 1998], monooxigenasa-2 que contiene flavina (FMO2) [J Biol Chem 273:30599, 1998], hidroxilasa de CMP-N-acetilneuramínico [Hayakawa et al., PNAS 98:11399, 2001] y el gen variable V10 del locus del receptor gamma de células T humanas [Zhang et al., Immunogenetics 43:196, 1996]. Se invita a los lectores a informarme sobre pseudogenes unitarios adicionales).
4.2. Pseudogenes duplicados clásicos. Existen muchos ejemplos de pseudogenes compartidos de este tipo; describiré solo uno. El gen de la esteroide 21-hidroxilasa codifica una enzima involucrada en el metabolismo de las hormonas esteroideas. En el ADN humano, la secuencia del gen de la 21-hidroxilasa, así como un gen adyacente que codifica "complemento C4", se ha duplicado; es decir, hay copias casi idénticas de segmentos de ADN situadas una al lado de la otra, cada copia conteniendo un gen de complemento C4 y una secuencia de esteroide 21-hidroxilasa. Sin embargo, solo la copia "B" del gen de la 21-hidroxilasa es funcional; la copia "A" en todos los humanos es un pseudogen, es decir, contiene múltiples mutaciones, incluida una deleción de 8 pb que impediría su función. La secuencia de la copia correspondiente "A" del chimpancé ha sido examinada; contiene la misma deleción incapacitante de 8 pb observada en el pseudogen humano (Kawaguchi, Am J Hum Genet 50:766-80, 1992).
Muchos de los pseudogenes centrómeros peculiares descritos anteriormente (en la sección 2.2.1.b) también están conservados en otros primates (Eichler et al., Human Molec Genet 5:899, 1996; Regnier et al, Human Molec Genet 6:9, 1997; Grewal et al., Gene 227: 79, 1999).
4.3. Pseudogenes procesados. Dado que el ADN humano puede contener aproximadamente cuatro veces más pseudogenes procesados que pseudogenes duplicados clásicos (extrapolando datos del cromosoma 22 [Dunham et al, Nature 402:489, 1999]), hay muchos más ejemplos de pseudogenes procesados (que pseudogenes clásicos) compartidos entre especies. Describiré uno que mis colegas y yo descubrimos: un pseudogén derivado del gen que codifica la inmunoglobulina epsilon, un tipo de anticuerpo que participa en reacciones alérgicas. En nuestros estudios destinados a investigar la base de la alergia, descubrimos una secuencia que se asemejaba al gen de la inmunoglobulina epsilon, excepto que carecía de intrones, presentaba múltiples mutaciones incapacitantes, tenía en su extremo una secuencia de casi continuas "A" (pareciendo un tracto de poli(A) ligeramente mutado) y estaba ubicado en un cromosoma diferente (cromosoma 9) al del gen funcional (cromosoma 14) (Max et al. Cell 29:691, 1982; Battey et al. PNAS 79:5956, 1982). Nuestra evidencia sugirió que este pseudogén procesado también existía en el ADN del chimpancé, y posteriores investigaciones detalladas de otros laboratorios (Kawamura y Ueda, Genomics 13:194, 1992) demostraron que pseudogenes casi idénticos existen en chimpancé, gorila, orangután y monos del Viejo Mundo. Al igual que en el caso de todas las inserciones de ADN compartidas por diferentes especies (véanse otros ejemplos a continuación), el argumento de que estas secuencias no fueron creadas independientemente sino que descienden de una inserción ancestral común se refuerza con la demostración de que las inserciones ocurrieron en la misma posición en el ADN de cada especie, es decir, el ADN que rodea la inserción es muy similar entre especies, tan cercano a idéntico como cabría esperar dada la ocurrencia de mutaciones que no son seleccionadas en contra.
4.4. SINEs. De las aproximadamente un millón de copias de secuencias Alu en el genoma humano, solo una pequeña fracción ha sido comparada entre humanos y otras especies de primates. Sin embargo, en varios segmentos largos de ADN donde se han obtenido las secuencias correspondientes en el ADN humano y de chimpancé, casi todas las secuencias Alu son compartidas entre estas dos especies. Por ejemplo, en el grupo de genes de globina ![]() mencionado anteriormente, las siete secuencias Alu encontradas en el ADN humano están presentes en el chimpancé, incrustadas exactamente en las mismas posiciones (Sawada et al. J Mol Evol 22:316, 1985). Lo mismo es cierto para las siete secuencias Alu cerca de un pseudogén derivado del gen de copia única cdc27hs (Gonzalez et al., Genomics 18:29, 1993).
mencionado anteriormente, las siete secuencias Alu encontradas en el ADN humano están presentes en el chimpancé, incrustadas exactamente en las mismas posiciones (Sawada et al. J Mol Evol 22:316, 1985). Lo mismo es cierto para las siete secuencias Alu cerca de un pseudogén derivado del gen de copia única cdc27hs (Gonzalez et al., Genomics 18:29, 1993).
Se han comparado las secuencias de muchos repetidores Alu en el ADN humano, permitiendo su clasificación en varias familias, basándose en el grado de similitud de la secuencia. Los miembros de ciertas familias se encuentran en el ADN de muchos primates diversos, mientras que otras familias parecen haberse dispersado más recientemente, ya que no son compartidas por otras especies. Se conocen varios ejemplos de inserciones de la familia "más joven" que son polimórficas en la población humana: es decir, ocurren en algunos individuos pero no en otros. De hecho, la frecuencia de ciertas inserciones Alu en diferentes poblaciones humanas se ha utilizado para deducir patrones probables de migración y mezcla génica en nuestros antepasados humanos. Tales observaciones son consistentes con la inserción de tales copias Alu después de la evolución humana. Además, la excelente salud de los individuos que carecen de ciertas inserciones Alu respalda la visión de que estas inserciones no cumplen ninguna función importante en la fisiología humana.
4.5. LINEs. Se han encontrado numerosas secuencias de LINE en la misma posición del ADN de humanos y otras especies, incluyendo ejemplos en el locus de las globinas, genes de pigmentos visuales y fosfatasa alcalina intestinal (revisado por Smit et al. J Mol Biol 246:401,1995). Algunos de los ejemplos reportados son compartidos por especies tan dispares como el humano y la vaca, lo que indica inserciones en antepasados mamíferos muy tempranos.
4.6. Retrovirus endógenos. Dado que los retrovirus endógenos son menos numerosos que las otras secuencias de ADN no funcionales discutidas aquí, y dado que una fracción relativamente pequeña de las secuencias de ADN humano conocidas han sido comparadas entre especies, existe una escasez de ejemplos de retrovirus endógenos compartidos. Sin embargo, se han reportado al menos cinco ejemplos diferentes de secuencias retrovirales casi idénticas incrustadas en la misma posición en el ADN humano y de chimpancé (Bonner et al. PNAS 79:4709, 1982; Dangel et al. Immunogenetics 42:41, 1995; Svensson et al Immunogenetics 41:74,1995; Medstrand & Mager J Virol 72:9782, 1998; Barbulescu et al. Curr Biol 9:861, 1999), todos aparentemente ejemplos de retrovirus que fueron "capturados" por los antepasados de nosotros hace millones de años. Se puede anticipar que se descubrirán ejemplos adicionales a medida que se vuelvan disponibles más datos de secuencia, especialmente desde el cromosoma Y, que ha sido descrito como un " cementerio" para secuencias de retrovirus endógenos tanto para humanos como para chimpancés (Kjellman et al. Gene 161:163, 1995).
4.7 Implicaciones de las secuencias sin función compartidas entre especies
Todos los ejemplos de secuencias sin función compartidas entre humanos y chimpancés refuerzan el argumento a favor de la evolución que sería convincente incluso si solo se conociera un ejemplo. Este argumento puede entenderse por analogía con los casos legales discutidos anteriormente en los que los errores compartidos fueron reconocidos como prueba de copia. La aparición del mismo "error"—es decir, el mismo pseudogén inútil o secuencia Alu o virus endógeno retrovirales en la misma posición en el ADN humano y de simio—no puede explicarse lógicamente por orígenes independientes de las dos secuencias. El argumento creacionista discutido anteriormente—que las similitudes en la secuencia de ADN simplemente reflejan los planes del creador para funciones proteicas similares en especies similares—no se aplica a secuencias que no tienen ninguna función para el organismo que las alberga. La posibilidad de que accidentes genéticos idénticos crearan el mismo dos pseudogén o Alu o virus endógeno retrovirales independientemente en dos especies diferentes por casualidad es tan improbable que puede ser descartada. Como en los casos de derechos de autor discutidos anteriormente, tales errores compartidos indican que debe haber ocurrido algún tipo de copia. Dado que no existe ningún mecanismo conocido por el cual las secuencias de simios modernos puedan ser copiadas en la misma posición del ADN humano o viceversa, la existencia de pseudogenes compartidos o retrotransposones lleva a la conclusión lógica de que tanto las secuencias humanas como las de simios fueron copiadas de secuencias ancestrales que debieron surgir en un ancestro común de humanos y simios.
Esta evidencia de un ancestro común cierra el argumento sobre la evolución humana/primates que se deriva de las secuencias compartidas sin función. Aunque los ejemplos documentados más numerosos de tales secuencias compartidas entre diferentes especies vinculan por casualidad a los humanos y a los simios (véase, por ejemplo, Hamdi et al, J Mol Biol 284:861, 1999), esto simplemente refleja el hecho de que el ADN de los humanos ha sido estudiado con mayor intensidad que el ADN de cualquier otra especie superior, mientras que se conoce una considerable secuencia homóloga de chimpancé. Es obvio, sin embargo, que la misma lógica idéntica podría utilizarse para vincular otras especies en diferentes ramas del árbol evolutivo, y se han reportado tales ejemplos, por ejemplo, SINEs que aclaran las relaciones entre especies de roedores (Furano J Biol Chem. 270: 25301, 1995; Verneau et al, PNAS 95: 11284, 1998) o que vinculan caballos a rinocerontes (Gallagher et al, Mamm Genome:140, 1999) o que establecen las afiliaciones filogenéticas de los tarsier (Schmitz et al., Genetics 157:777, 2001) o rumiantes pecorinos (Nijman et al J Mol evol 54:9, 2002). Especies tan dispares como humanos y ratones han sido vinculadas por ejemplos de la antigua familia de SINE conocida como MIRs (Repeticiones Intercaladas Mamíferas Amplias; véase Smit y Riggs, Nucleic Acids Research 23:98, 1995; Jurka et al, Nucleic Acids Research 23:170, 1995) que se encontraron incrustadas en la ubicación homóloga en los genes de mioglobina y N-myc humanos y murinos (Donehower, Nucleic Acids Research 17:699, 1989; obsérvese que en el momento de esta descripción la secuencia conservada no se reconocía como un SINE). Además, las inserciones antiguas de LINE vinculan a los humanos con la vaca, como se mencionó anteriormente (inserciones de LINE similares aguas arriba de los genes de fosfatasa alcalina intestinal en ambas especies), así como con la rata (inserciones de LINE similares en el primer intrón de la subunidad alfa2 de los genes de ATPasa sodio-potasio; Smit et al, J Mol Biol 246:401, 1995) y con el ratón (por ejemplo, inserciones de LINE en la región mnd2 del cromosoma 2p13 [Jang et al, Genome Res 9:51, 1998] y cerca del gen CD4 en el cromosoma 12p13 humano [Ansari-Lari et al Genome Res 8:29, 1998]). Con comparaciones adicionales de secuencias de largas tramas homólogas de ADN humano y de ratón anticipadas del Proyecto Genoma Humano y del Proyecto Genoma del Ratón, es probable que se descubran secuencias adicionales de LINE compartidas entre estas especies.
Un ejemplo particularmente impresionante de retrotransposones compartidos ha sido recientemente reportado, vinculando a los cetáceos (ballenas, delfines y marsopas) a los rumiantes y a los hipopótamos, y es instructivo considerar este ejemplo en algún detalle. Los cetáceos son animales que viven en el mar que presentan similitudes importantes con los mamíferos que viven en tierra; en particular, las hembras tienen glándulas mamarias y amamantan a sus crías. Los científicos que estudian la anatomía y la fisiología de los mamíferos han demostrado las mayores similitudes entre los cetáceos y el grupo de mamíferos conocido como artiodáctilos (ungulados de pezuña par) que incluyen vacas, ovejas, camellos y cerdos. Estas observaciones han llevado a la visión evolucionista de que las ballenas evolucionaron de un ancestro artiodáctilo de cuatro patas que vivía en tierra. Los creacionistas se han aprovechado de las diferencias obvias entre los artiodáctilos familiares y las ballenas, y han ridiculizado la idea de que las ballenas podrían haber tenido ancestros de cuatro patas que vivían en tierra. Los creacionistas que afirman que los cetáceos no surgieron de mamíferos terrestres de cuatro patas deben ignorar o de alguna manera descartar la evidencia fósil de aparentes ancestros de ballenas que se ven exactamente como uno prediría para especies transicionales entre mamíferos terrestres y ballenas: con patas diminutas y con estructuras de oído intermedias entre las de los artiodáctilos modernos y los cetáceos (Nature 368:844,1994; Science 263: 210, 1994). (Una discusión sobre especies fósiles de ancestros de ballenas con referencias puede encontrarse en http://www.talkorigins.org/faqs/faq-transitional/part2b.html#ceta) Los creacionistas también deben ignorar o descartar la evidencia que muestra la gran similitud entre las secuencias génicas de los cetáceos y los artiodáctilos (Molecular Biology & Evolution 11:357, 1994; ibid 13: 954, 1996; Gatesy et al, Systematic Biology 48:6, 1999).
Recientemente, la evidencia de retroposones ha consolidado la relación evolutiva entre las ballenas y los artiodáctilos. Shimamura et al. (Nature 388:666, 1997; Mol Biol Evol 16: 1046, 1999; véase también Lum et al., Mol Biol Evol 17:1417, 2000; Nikaido y Okada, Mamm Genome 11:1123, 2000) estudiaron secuencias SINE altamente reduplicadas en el ADN de todas las especies de cetáceos examinadas. Estos SINE también se encontraron presentes en el ADN de los rumiantes (incluyendo vacas y ovejas), pero no en el ADN de camellos, cerdos o mamíferos más distantes como el caballo, el elefante, el gato, el humano o el canguro. Estos SINE aparentemente originaron en una rama específica de los artiodáctilos ancestrales después de que esta rama se separara de los camellos, cerdos y otros mamíferos, pero antes de la divergencia de las líneas que conducen a los cetáceos modernos, los hipopótamos y los rumiantes. (Véase la Figura 5.) En apoyo de este escenario, Shimamura et al. identificaron dos inserciones específicas de estos SINE en el ADN de ballenas (inserciones B y C en la Figura 5) y demostraron que en el ADN de hipopótamo, vaca y oveja, estos mismos dos sitios contenían los SINE; pero en el ADN de camello y cerdo, los mismos sitios estaban "vacíos" de inserciones. Más recientemente, se ha identificado al hipopótamo como el pariente terrestre vivo más cercano de los cetáceos, ya que los hipopótamos y las ballenas comparten inserciones de retroposones (ilustradas por D y E en la Figura 5) que no se encuentran en ningún otro artiodáctilo (Nikaido et al, PNAS 96:10261, 1999). La estrecha relación entre hipopótamo y ballena es consistente con las comparaciones de similitud de secuencias previamente reportadas (Gatesy, Mol Biol Evol 14:537, 1997) y con los hallazgos fósiles recientes (Gingerich et al., Science 293:2239, 2001; Thewissen et al., Nature 413:277, 2001) que resuelven conflictos paleontológicos anteriores con la estrecha relación entre ballenas y hipopótamos. (Algunos lectores se han preguntado: si los rumiantes están más estrechamente relacionados con las ballenas que con los cerdos y camellos, ¿por qué los rumiantes son anatómicamente más similares a los cerdos y camellos que a las ballenas? Aparentemente, esto resulta del hecho de que los rumiantes, cerdos y camellos cambiaron relativamente poco desde su último ancestro común, mientras que la línea de los cetáceos cambió drásticamente al adaptarse a un estilo de vida acuático, eliminando así muchas de las características —como pezuñas, pelo y patas traseras— que se comparten entre sus parientes rumiantes cercanos y los cerdos y camellos más distantes. Este escenario ilustra el hecho de que el desarrollo evolutivo rápido de adaptaciones a un nuevo nicho puede ocurrir a través de mutaciones funcionales clave, dejando la mayor parte del ADN relativamente sin cambios. La estrecha relación particular entre ballenas y hipopótamos es consistente con varias adaptaciones compartidas a la vida acuática, incluyendo el uso de vocalizaciones subacuáticas para la comunicación y la ausencia de pelo y glándulas sebáceas.) Por lo tanto, la evidencia de retroposones apoya fuertemente el origen de las ballenas de un ancestro común de hipopótamos y rumiantes, consistente con la interpretación evolutiva de los fósiles y las similitudes generales de secuencias de ADN. De hecho, la lógica de la evidencia de SINE compartidos es tan poderosa que los SINE pueden ser los caracteres disponibles más adecuados para deducir la parentescos de las especies (Shedlock y Okada, Bioessays 22:148, 2000), incluso si no son perfectos (Myamoto, Curr. Biology 9:R816, 1999).
|
|
|
Figura 5. Las inserciones específicas de SINE pueden actuar como "trazadores" que iluminan las relaciones filogenéticas. Esta figura resume algunos de los datos sobre SINEs encontrados en artiodáctilos vivos y muestra cómo las inserciones compartidas pueden interpretarse en relación con la ramificación evolutiva. Un evento de inserción específico de SINE ("A" en la Figura) parece haber ocurrido en un ancestro común primitivo de cerdos, rumiantes, hipopótamos y cetáceos, ya que esta inserción está presente en estos descendientes modernos de ese ancestro común; pero está ausente en camellos, que se separaron de las otras especies antes de que se insertara este SINE. Inserciones más recientes B y C están presentes solo en rumiantes, hipopótamos y cetáceos. Las inserciones D y E son compartidas solo por hipopótamos y cetáceos, identificando así al hipopótamo como el pariente vivo más cercano de los cetáceos (al menos entre las especies examinadas en estos estudios). Las inserciones de SINE F y G ocurrieron en la línea de los rumiantes después de que se separó de las otras especies; y las inserciones H e I ocurrieron después de la divergencia de la línea de los cetáceos. |

Aunque algunos creacionistas aceptan la evidencia de la selección natural de variantes menores (por ejemplo, la divergencia de las pinzuelas de Darwin en las islas Galápagos), a las que llaman "microevolución", la mayoría de los creacionistas niega que la evolución pueda explicar cambios más significativos, a los que designan como "macroevolución". Sin embargo, los pseudogenes/retrotransposones compartidos descritos aquí proporcionan una fuerte evidencia de que los humanos comparten ancestros comunes con especies tan dispares como monos, vacas y ratones. Por lo tanto, aunque quizás carezcamos de evidencia convincente de que cualquier fósil en particular sea ancestral de una especie moderna específica, y aunque no tengamos evidencia fósil que identifique claramente al último ancestro común entre humanos y vacas o entre ballenas y rumiantes, podemos estar seguros, a partir de los errores compartidos descritos aquí, de que estas especies ancestrales comunes existieron. Esta conclusión a su vez implica que las características novedosas significativas (por ejemplo, la marcha bípeda humana y el desarrollo cerebral, y las adaptaciones de los cetáceos a la vida acuática) deben haberse desarrollado entre el tiempo en que vivieron los respectivos ancestros comunes y la actualidad. Estos cambios son claramente extensos lo suficienteos como para ser llamados "macroevolución", por lo que el "argumento de los errores compartidos" es una evidencia poderosa para la macroevolución. Esta conclusión parece sólida, ya que no se ha propuesto en la literatura científica ninguna explicación alternativa de estos errores compartidos consistente con el origen independiente de estas especies animales.
Claramente, el argumento de los "errores compartidos" proporciona una fuerte evidencia de cambios macroevolutivos en la evolución de los mamíferos, y por lo tanto refuta una posición creacionista comúnmente sostenida. Pero para ser justos, debemos aclarar que este argumento no compra todo el juego evolucionista. Aunque la evidencia de errores compartidos implica la descendencia común de diversas especies de mamíferos, no aborda si estas especies evolucionaron de sus últimos ancestros comunes a través de los mecanismos darwinianos de mutación y selección natural o a través de otros mecanismos alternativos. Otra limitación es que no hay ejemplos de "errores compartidos" que vinculen a los mamíferos con otras ramas del árbol genealógico de la vida en la Tierra. Por ejemplo, aunque especies tan diversas como gusanos, levaduras y plantas tienen elementos LINE en sus genomas, no se han reportado ejemplos de inserciones específicas de LINE en posiciones homólogas entre ningún mamífero y no mamífero, según mi conocimiento (aunque bienvenido sea el aporte de los lectores sobre este punto). Tales ejemplos podrían esperarse que sean difíciles de encontrar, ya que se cree que los últimos ancestros comunes de los mamíferos y los reptiles vivieron hace más de 200 millones de años, tiempo suficiente para que las similitudes de secuencia que una vez existieron en ADN sin función como pseudogenes y retrotransposones hayan sido en gran parte borradas por la acumulación de numerosas mutaciones. Por lo tanto, las relaciones evolutivas entre ramas distantes en el árbol genealógico evolutivo deben basarse en otra evidencia además de los "errores compartidos". (Dicha evidencia podría incluir otros "cambios genómicos raros" (RGCs) además de la inserción de retrotransposones, como la inserción o delección de secuencias codificantes o intrones (por ejemplo, Venkatesh et al., PNAS 98:11382, 2001), translocaciones cromosómicas e inversiones reveladas por citogenética comparativa, y variantes en el código genético, todo resumido en Rokas y Holland, Trends Ecol & Evol 15:454, 2000. La parentescía de las especies también puede inferirse de árboles de similitud de secuencia tradicionales basados en comparaciones de los genes correspondientes de diferentes especies.) Como una limitación final y bastante obvia del argumento de los "errores compartidos", debe quedar claro que este argumento no tiene que ver con las cuestiones del origen de la vida, que los creacionistas comúnmente agrupan con la evolución.
5. Respuestas de los creacionistas al argumento de las secuencias compartidas sin función
Los creacionistas tienden a evitar mencionar el argumento presentado en este ensayo, ya que proporciona evidencia persuasiva para la evolución, pero el portavoz creacionista Duane Gish ha comentado sobre el argumento cuando ha sido confrontado con él en debates; y han aparecido algunas otras discusiones creacionistas sobre pseudogenes. Examinemos primero varias de las respuestas del Dr. Gish.
5.1 Algunos "pseudogenes" procesados son funcionales, por lo que podrían ser ejemplos de "diseño similar para función similar."
Como se mencionó anteriormente (2.2.1.c), las copias retrotranscritas de transcripciones de ARN de genes pueden, raramente, insertarse en el ADN cerca de un promotor existente o de alguna otra manera que permita su transcripción de una forma útil para el organismo. Tales copias (que son realmente genes procesados en lugar de pseudogenes procesados) pueden, por lo tanto, proporcionar alguna función que ejerce presión selectiva contra mutaciones debilitantes. Se han reportado varios ejemplos de esta posibilidad, como se mencionó anteriormente en la sección 2.2.1.c; y estos podrían interpretarse como "diseño similar para función similar". Pero estos ejemplos comparten una característica que claramente los distingue de los cientos de ejemplos de pseudogenes procesados inútiles reportados: carecen de mutaciones debilitantes que impedirían la función, y por lo tanto permanecen capaces de codificar una proteína útil. Entre los pseudogenes procesados auténticos —es decir, copias de genes retroposados con múltiples mutaciones debilitantes como codones de parada— no se han reportado ejemplos con función documentada [Ver nota añadida a continuación]. (Los lectores que crean que existen ejemplos que contradicen esta declaración son invitados a contactarme con las referencias bibliográficas; modificaré este artículo según sea necesario). Así, el argumento del Dr. Gish simplemente refleja su errónea agrupación de dos clases distintas de copias de genes retroposados: genes procesados y pseudogenes procesados. Y el Dr. Gish aún no ha ofrecido ningún argumento que explique —en términos de función diseñada inteligentemente— los numerosos ejemplos de secuencias retroposadas compartidas que, a diferencia de los pseudogenes, ni siquiera derivan de ADN que tenga un papel funcional.
Nota añadida el 5 de mayo de 2003: Acaba de publicarse un ejemplo de un pseudogén con función documentada. Una secuencia llamada Makorin1-p1 no codifica una proteína funcional, pero su transcripción de ARNm estabiliza el ARNm del gen funcional Makorin1 del cual aparentemente se derivó (Hirotsune et al, Nature 423:91,2003). El conocimiento actualmente disponible de la literatura publicada no sugiere roles funcionales para pseudogenes procesados además de Makorin1-p1. Es posible que estudios futuros descubran otros pseudogenes con aparente función (pero véase la siguiente sección).
5.2 Algunos órganos que anteriormente se consideraban vestigiales han sido encontrados más recientemente que tienen función; sabemos demasiado poco sobre estas nuevas características de ADN para estar seguros de que no se descubrirá una función para ellas en el futuro.
Imaginen a un acusado en un juicio por asesinato defendiéndose contra abrumadoras pruebas incriminatorias con el argumento paralelo de que, ya que algunos criminales condenados han sido posteriormente absueltos, él (el acusado actual) debería ser absuelto ahora, porque en algún momento del futuro podría encontrarse evidencia que lo aclare. Esta defensa sería tan ridícula como el argumento del Dr. Gish. Los científicos (y los jurados) deben sacar sus conclusiones basándose en la mejor evidencia disponible en ese momento. Es cierto que la evidencia posterior puede absolver a un criminal condenado o invalidar una teoría científica. Esta posibilidad debería fomentar la humildad y advertirnos contra conclusiones dogmáticas (y quizás contra la pena de muerte); pero no debería disuadirnos de sacar las conclusiones más razonables de los datos disponibles. Nuestro conocimiento actual respalda la interpretación de que la mayoría de los pseudogenes/retrotransposones compartidos son evidencia de la descendencia común y la macroevolución. Si en el futuro—para una secuencia particular de Alu o LINE-1 o retrovirus endógeno compartida entre humanos y otra especie—se descubre evidencia de función, entonces esta secuencia particular podría interpretarse razonablemente por el paradigma creacionista de "secuencia similar diseñada para función similar"; y por lo tanto, este retrotransposón tendría que ser eliminado de la lista de secuencias sin función compartidas que ofrecen evidencia de la evolución. Los cientos de miles de ejemplos restantes en esta lista continuarían ofreciendo un apoyo válido para la evolución.
Además, aunque estas secuencias de ADN vestigiales se descubrieron más recientemente que los órganos vestigiales conocidos en el tiempo de Darwin, sabemos lo suficiente sobre cómo surgen que no necesitamos postular ningún diseñador misterioso o función desconocida para explicarlas. Sabemos que los requisitos previos para la formación de SINEs y otros retrotransposones —es decir, transcripciones de ARN y transcriptasa inversa— están presentes en niveles bajos en células germinales estudiadas en el laboratorio, donde serían capaces, sin ninguna intervención sobrenatural, de generar retrotransposones que podrían transmitirse a generaciones futuras. Este hecho predeciría que las inserciones de retrotransposones deben estar ocurriendo a cierta frecuencia incluso hoy en día. De hecho, se han documentado inserciones específicas de elementos Alu en el ADN de individuos vivos. Por ejemplo, se encontró un elemento Alu insertado en el ADN de un paciente con neurofibromatosis tipo I, dañando el gen asociado con esta enfermedad (Wallace et al. Nature 353:6347, 1991). El padre y la madre del paciente tenían copias intactas del gen sin inserción de Alu, por lo que la inserción debe haber ocurrido en las células germinales de uno de los padres o muy temprano en el desarrollo embrionario del paciente. De manera similar, se encontró que un elemento LINE recién insertado había dañado el gen para una proteína de coagulación sanguínea, causando hemofilia en otro paciente cuyos padres carecían de esta inserción (Kazazian et al. Nature 332:164, 1988). (Otros ejemplos de inserción de LINE o Alu que causan enfermedades son revisados por Kazazian [en Curr Opin Genet & Devel 8:343, 1998], por Miki [Human Genetics 43:77, 1998] y por Deininger y Batzer [Molec Genet & Metab 6:183, 1999].) Se estima que los nuevos eventos de retroposición ocurren en el 1% al 10% de la población humana (Kazazian Nature Genet 22:130, 1999). Carlton et al (Mamm Genome 6:90, 1995) observaron la aparición de novo de un pseudogen procesado cuando proporcionaron una fuente de transcriptasa inversa infectando células cultivadas con un retrovirus; mientras que Esnault et al. (Nature Genet 24:363, 2000) y Wei et al. (Molec Cell Biol 21:1439, 2001) observaron la formación de pseudogenes procesados resultante de la RT de un elemento LINE humano. Utilizando un ensayo sensible para detectar retroposición, Maestre et al. (EMBO J 14:6388, 1995) fueron capaces de detectar copias retroposadas de una secuencia de gen marcada siendo insertadas en el ADN de células humanas mientras crecían en el laboratorio incluso sin la adición de transcriptasa inversa exógena. Además, Jensen y Heidmann (EMBO J 10:1927, 1991) detectaron la retroposición continua de una copia marcada de LINE en Drosophila.
Recientemente, Feng et al. (Cell 87: 905, 1996) demostraron que la enzima transcriptasa inversa activa codificada por una copia recién insertada de LINE tiene una actividad adicional e inesperada: es una endonucleasa, es decir, es capaz de provocar cortes en el ADN que podrían servir como puntos de inserción para nuevos eventos de retroposición. De hecho, esta endonucleasa corta el ADN con características de secuencia particulares, y las mismas características fueron observadas en las posiciones de inserción de varias copias de LINE seleccionadas al azar del ADN humano. (Véase también Cost y Boeke, Biochemistry 37:18081, 1998). Este resultado sugiere que las secuencias de LINE están tan bien adaptadas para la replicación "egoísta" en el genoma que no dependen de cortes aleatorios generados en el ADN para sus inserciones, sino que generan sus propios cortes. Para probar esta idea, Moran et al. (Cell 87:917, 1996; véase también Ostertag et al., Nucl Ac Res 28:1418, 2000) construyeron una secuencia de LINE diseñada de tal manera que si generaba nuevas copias retropuestas en cualquier célula, estas células podrían seleccionarse y contarse. Cuando esta secuencia se introdujo en células de cultivo de tejido humano, se produjeron rutinariamente nuevas copias retropuestas. Al probar los efectos de mutaciones en varios segmentos de la secuencia de LINE, se demostró que la retroposición eficiente requería tanto la actividad de transcriptasa inversa como la actividad de endonucleasa presentes en la misma proteína. (Esta proteína también es necesaria para la formación eficiente de pseudogenes procesados inducidos por LINE [Esnault et al. Nature Genet 24:363, 2000]).
Observaciones como estas refuerzan la noción de que las secuencias de retroposones que observamos en nuestro ADN y en el ADN de otros mamíferos no fueron creadas por fuerzas misteriosas que actuaron únicamente en el pasado antiguo con propósitos inescrutables, sino por simples accidentes genéticos que ocurren a baja frecuencia como resultado de peculiaridades de la bioquímica celular, y que no sirven para ningún propósito. El hecho de que muy pocos de estos accidentes genéticos puedan crear alguna función beneficiosa (Britten RJ PNAS 93:9374,1996; Britten RJ Gene 205:177,1997) no debilita en absoluto esta interpretación; tales eventos son simplemente ejemplos de mutaciones beneficiosas raras cuya ocurrencia constituye la base para el cambio evolutivo adaptativo y cuya existencia parece tan difícil de aceptar para los creacionistas. Como ocurre con la mayoría de las mutaciones, la abrumadora mayoría de las inserciones de retroposones ocurren en el ADN no funcional entre genes y no tienen efecto sobre la célula u organismo; y es este vasto conjunto de inserciones, compartido entre especies, lo que proporciona la base para el argumento actual que apoya la evolución.
5.3 Si todas estas secuencias realmente fueran no funcionales, habrían sido eliminadas a lo largo del tiempo evolutivo.
Este argumento refleja ignorancia de los hechos discutidos anteriormente en la sección 3. Para repetir: no se conoce ningún mecanismo mediante el cual las secuencias de ADN no funcionales podrían distinguirse de las funcionales y ser seleccionadas para su eliminación por enzimas celulares. Las bacterias parecen estar bajo presión selectiva para eliminar el ADN no funcional; los cromosomas bacterianos tienen muy poco ADN entre genes, quizás porque la competencia bajo condiciones de crecimiento rápido puede favorecer cromosomas que se replican rápidamente —es decir, los más cortos— y, por lo tanto, pueden seleccionar células que hayan eliminado cualquier ADN no funcional. Pero no hay evidencia de tal presión selectiva para los cromosomas mamíferos, en los que los genes están ampliamente separados entre sí y en los que las regiones no funcionales constituyen aparentemente el 90-95% del ADN. De hecho, uno podría preguntar: ¿por qué entonces nuestros cromosomas no están repletos de secuencias de retroposones a una frecuencia aún mayor que la realmente observada? Una respuesta razonable es que nuestros antepasados estaban bajo presión selectiva para suprimir la retroposición, ya que altas frecuencias de inserción de retroposones aumentarían la tasa de daño genético causado por inserciones incapacitantes en genes. Además, es concebible que una fracción mayor de nuestro ADN haya originado a través de la retroposición de lo que ahora podemos reconocer; algunos pseudogenes o inserciones de retroposones muy antiguos pueden haber sufrido tantas mutaciones aleatorias desde su inserción que sus identidades como pseudogenes o retroposones han sido borradas. Sin embargo, a la tasa de mutación estimada para secuencias no seleccionadas, la obliteración completa de un retroposón típico por mutaciones requeriría más de 100 millones de años. Por lo tanto, no sería sorprendente para un evolucionista que las secuencias de retroposones sin función que se insertaron en un ancestro común de humanos y vacas aún puedan ser detectables mediante comparaciones computarizadas de secuencias de ADN.
5.4 Se han encontrado roles importantes para regiones de ADN que anteriormente se consideraban sin función
En un reciente debate con el Dr. Gish, citó una reseña en Science titulada "Mining treasures from 'junk' DNA" (263:608, 1994), pareciendo implicar que esta reseña sugiere funciones para pseudogenes y retroposones que serían consistentes con la visión creacionista de que fueron diseñados para funcionar de manera similar en especies similares. De hecho, esta reseña discute evidencia sobre posibles funciones de secuencias repetitivas centroméricas y teloméricas, minisatélites, intrones y regiones no traducidas 3'. Menciona pseudogenes y retroposones pero no hace ninguna sugerencia de que estos elementos particulares tengan función, por lo que esta reseña no ofrece ningún argumento contra los puntos planteados en este ensayo. No obstante, dado que ha habido otras especulaciones sobre posibles funciones para el ADN fuera de las secuencias de codificación génica, vale la pena considerar por qué los científicos generalmente aceptan la noción de que la mayor parte de este ADN es basura.
Primero, sabemos varios mecanismos mediante los cuales la longitud del ADN puede aumentar a través de accidentes genéticos como las duplicaciones del ADN e la inserción de retrotransposones, los cuales han sido observados en el laboratorio o ocurren en humanos sin efectos aparentes; por lo tanto, es razonable suponer que estos mecanismos operaron en el pasado para aumentar el tamaño del genoma sin afectar la función. Parece haber poca o ninguna presión selectiva para reducir el tamaño de los genomas nucleares de los vertebrados; y no existe un mecanismo aparente para eliminar selectivamente el ADN inútil. Las grandes deleciones que eliminan ADN funcional son seleccionadas en contra. Estas observaciones predecirían la acumulación de ADN inútil como resultado de accidentes genéticos aleatorios, por lo que cuando vemos ADN que parece no funcional, no deberíamos asumir necesariamente que tiene una función que no entendemos.
En segundo lugar, cuando se compara la secuencia de ADN entre especies como la humana versus la del ratón, las secuencias que se sabe que tienen función —en particular, las secuencias codificantes de genes— se encuentran altamente similares, lo cual es consistente con la presión selectiva que elimina a los individuos que tienen mutaciones deletéreas en estas regiones funcionales. Por el contrario, las regiones de ADN sin función conocida —por ejemplo, secuencias no codificantes entre genes— generalmente se comportan como si no estuvieran bajo ninguna presión selectiva, es decir, aparentemente acumulan mutaciones a una tasa mucho más alta, por lo que hay poca conservación de secuencias entre especies distantly relacionadas. Como una excepción que pone a prueba la regla, las comparaciones de secuencias no codificantes entre especies ocasionalmente detectan "islas" de secuencias conservadas cortas en regiones no codificantes. Algunas de estas han resultado corresponderse con regiones reguladoras como elementos promotor o potenciador que controlan cuándo se expresa un gen cercano. Un ejemplo de tal "isla" conservada entre conejo, ratón y humano fue descubierta en mi propio laboratorio [Emorine et al., Nature 304:447, 1983]; resultó representar un potenciador importante. Estos tipos de regiones reguladoras generalmente ocupan mucho menos ADN que las secuencias codificantes de los genes que regulan, por lo que no pueden representar una función probable para la mayoría del ADN no codificante. La buena correlación entre función y conservación de secuencias apoya la idea de que la mayoría de las secuencias mal conservadas no tienen función. Sin embargo, debe notarse que para la mayoría de las "islas" de secuencias conservadas en el ADN entre genes (Shabalina et al., Trends Genet 17:373, 2001), aún no se ha descubierto ninguna función. Algunas pueden incluir especies de ARN que funcionan sin ser traducidas a proteína.
Un tercer argumento, aunque relacionado, se deriva de la observación de que la inserción de un retroposón en una secuencia funcional es una manera potente de destruir esa función. Ejemplos de inserciones que ocurren naturalmente se discutieron en la sección 5.2 anterior; y la inserción intencional de retroposones está siendo ampliamente utilizada como una herramienta de laboratorio para crear paneles de cepas de ratón, drosophila o levadura con diferentes funciones génicas destruidas. Sin embargo, la mayoría de los ejemplos de inserciones de retroposones entre genes no tienen ningún efecto aparente en los individuos que los portan; por ejemplo, las secuencias Alu que son polimórficas en el ADN humano parecen ser inofensivas cuando están presentes. Por lo tanto, es razonable inferir que estas inserciones no interrumpieron ninguna secuencia funcional. (Por supuesto, es imposible descartar la posibilidad formal de que algunas secuencias funcionales hipotéticas fuera de los genes puedan seguir funcionando a pesar de la presencia de una inserción de retroposón.)
Finalmente, se conocen varios ejemplos de pares de especies que tienen una complejidad aparente similar pero tamaños de genoma muy diferentes (paradoja del valor C). El pez globo Fugu tiene aproximadamente una cuarta parte del tamaño del genoma de otras especies de peces, pero aproximadamente el mismo número de genes. La principal diferencia es una menor cantidad de ADN entre genes en el ADN de Fugu (por ejemplo, véase Elgar et al. Genome Res 9:960, 1999). Aunque permanecen preguntas sobre la interpretación de esta diferencia, parece que gran parte del ADN entre genes en la mayoría de los genomas de peces (y probablemente en el nuestro también) es prescindible. (Por el contrario, las pequeñas regiones de secuencia no codificante que se conservan entre Fugu y Homo frecuentemente corresponden a secuencias regulatorias funcionales.)
Es imposible probar la ausencia de función para cualquier región de ADN. Además, es probable que se encuentre alguna función para algunas regiones cortas adicionales de ADN no codificante que actualmente no se reconocen como funcionales. No obstante, como se indicó anteriormente, los científicos concluyen de manera provisional basándose en los datos disponibles actualmente en lugar de en posibilidades hipotéticas de datos futuros; y los argumentos que acabo de presentar basados en la evidencia disponible sugieren que la mayoría de las secuencias de ADN que parecen carecer de función son simplemente eso.
5.5 Los pseudogenes cumplen una función: proporcionan una copia de "respaldo" que puede corregirse para codificar una proteína útil si el gen funcional sufre una mutación crítica.
El Dr. Gish no proporcionó especificaciones para esta afirmación, pero quizás se refería a una sugerencia reciente de que un pseudogén de ribonucleasa seminal bovina fue recientemente "corregido" para volver a ser funcional mediante un proceso conocido como "conversión génica" (Trabesinger-Ruef et al. FEBS Lett 382:319, 1996). Aunque esto puede ocurrir ocasionalmente, se han descrito en la literatura muchos más casos en los que defectos en un pseudogén inducen mutaciones dañinas en un gen funcional cercano mediante conversión génica, inactivando el gen funcional. Como ejemplo de este tipo de evento, en casi todos los pacientes que sufren de deficiencia en la 21-hidroxilasa de los esteroides porque su copia (normalmente) funcional del gen 21-hidroxilasa "B" ha sido inactivada por mutaciones puntuales, estas mutaciones aparentemente resultaron de la conversión génica por la copia del pseudogén "A" (Collier et al, Nat Genet 3:260, 1993; Carrera et al. Hum Hered 43:190, 1996). Se cree que conversiones génicas similares por un pseudogén han inactivado el gen glucocerebrosidasa en pacientes con enfermedad de Gaucher (Eyal et al. Gene 96:277, 1990), el gen 14.1 que codifica una "cadena ligera sustituta" de inmunoglobulina en un paciente con inmunodeficiencia (Minegishi et al., J Exp Med 187:77, 1998), el gen ABCC6 en pacientes con pseudoxantoma elástico (Cai et al, (J Mol Med 79:536,2001), el gen PKD1 en la poliquistosis renal autosómica dominante (Watnick et al. Hum Mol Genet 7:1239, 1998), el gen beta-cristalina B2 en la catarata hereditaria autosómica dominante (Vanita et al. J Med Genet 38:392,2001), el gen NCF1 en la enfermedad granulomatosa crónica (Vazquez et al., Exp Hematol 29:234,2001) y el gen del factor von Willebrand en pacientes con enfermedad de von Willebrand (Eikenboom et al., PNAS 91:2221, 1994), para nombrar solo algunos ejemplos. En otros casos, los eventos de conversión génica aparentemente han transferido información genética entre dos pseudogenes (Shapiro y Moshirfar, J Mol Biol 209:181, 1989) o entre dos genes funcionales (Ollo y Rougeon, Cell 32:515, 1983). Dado que se ha reportado que la conversión génica que involucra pseudogenes ocurre con efectos dañinos o neutros más que con efectos beneficiosos, la hipótesis de que los pseudogenes fueron "diseñados" con el potencial de la conversión génica como su propósito parece poco convincente. (El único ejemplo donde las copias de pseudogenes claramente cumplen una función importante al transferir su secuencia a otra copia génica mediante conversión génica ocurre en la diversificación somática de los genes de la región variable de inmunoglobulinas de pollos y conejos; de las muchas mutaciones que se generan mediante este mecanismo, aquellas pocas que proporcionan un "ajuste mejor" entre la inmunoglobulina y su antígeno objetivo son seleccionadas para su expresión. Esta selección de una función mejorada entre genes que han experimentado cambios de secuencia cuasi-aleatorios es un modelo biológico atractivo para las mejoras evolutivas en la función de las proteínas. Irónicamente, en varias debates conmigo, el Dr. Gish negó que ocurra tal diversificación somática, aunque claramente estaba totalmente ignorante sobre la literatura científica concerniente a los genes de anticuerpos.) Además de ser poco convincente por la razón descrita anteriormente, la idea del Dr. Gish de que los pseudogenes fueron creados para proporcionar una copia génica de "respaldo" no ofrece ninguna explicación creacionista para los retroposones compartidos más numerosos que no son pseudogenes.
5.6 Todo este asunto de los retrotransposones es realmente demasiado difícil de entender.
El Dr. Gish utilizó este recurso de apelación a la audiencia en un debate reciente con mí. Pareció estar instando a la audiencia a ignorar las implicaciones del argumento de los pseudogenes compartidos y a desestimar el hecho de que él (el Dr. Gish) no podía encontrar contraargumentos válidos para oponerse a él. Esto es una maniobra de debate típica para los creacionistas: usar el humor o la invocación de la fe o alguna otra apelación irrelevante para distraer a una audiencia no especializada de darse cuenta de que una posición creacionista ha sido efectivamente refutada.
(Este ensayo fue enviado al Dr. Gish para solicitar cualquier argumento adicional contra los puntos presentados aquí. No se recibió respuesta.)
5.7 Además del Dr. Gish, el creacionista John Woodmorappe ha comentado sobre los pseudogenes (Noah's Ark, a Feasibility Study, 1996, publicado por ICR, p. 202; Bible-Science News 33:7, 1995). Presenta varios de los mismos argumentos que el Dr. Gish (véase 5.2 y 5.4 más arriba), pero añade algunos propios. La interpretación creacionista de los pseudogenes ofrecida por Woodmorappe es que algunos pseudogenes pueden ser "el resultado de cambios degenerativos en los organismos vivos desde la Caída". Esta interpretación parece plausible, y—si ignoramos la parte de la "Caída"—no es muy diferente de la idea evolutiva de que los pseudogenes surgen por accidentes genéticos aleatorios. Sin embargo, esta interpretación ignora completamente el hecho de que muchos pseudogenes son compartidos entre los simios y los humanos, ubicados en las mismas posiciones y compartiendo los mismos defectos genéticos, aparentemente el resultado del mismo accidente genético o "cambio degenerativo" en un ancestro común. (Si estos pseudogenes compartidos surgieron después de la "Caída" como sugiere Woodmorappe, ¿ocurrió acaso la "Caída" antes de que el hombre se separara de los simios?)
5.8 Al abordar los pseudogenes compartidos, Woodmorappe intenta oscurecer su fuerte apoyo a la evolución al afirmar que para ciertos pseudogenes, el grado de "parentesco" inferido a partir de la presencia o ausencia del pseudogene en diferentes especies contradice el "parentesco" de las especies inferido por los evolucionistas a partir de otras características. En este argumento, Woodmorappe se alinea con otros argumentos creacionistas que nos invitan a descartar la evolución debido a casos específicos que violan una interpretación simplista de la evolución, y a ignorar el vasto número mayor de ejemplos que apoyan la evolución. En la larga y compleja historia de la vida en la Tierra, se han generado muchas excepciones a nociones simplistas, por ejemplo, casos donde fósiles más antiguos yacen por encima de los más jóvenes (debido al plegamiento de estratos geológicos o fallas de empuje) o ejemplos donde las similitudes de secuencias de pequeños tramos de ADN comparados entre especies parecen violar las relaciones aceptadas (debido a errores estadísticamente esperados debido a muestras pequeñas). De manera similar, podemos esperar casos en los que un pseudogene o retroposón que surgió en el ancestro de tres especies modernas (A, B y C) pueda ser eliminado en una de ellas (digamos C), sugiriendo una relación más cercana entre A y B de la que está justificada por otros fundamentos. Un ejemplo como este no debería hacernos descartar lo que aprendemos de la mayoría de los pseudogenes y retroposones compartidos; por el contrario, deberíamos tener precaución al sacar generalizaciones de casos excepcionales.
| Caja 3 |
|---|
|
Woodmorappe describe un ejemplo de un pseudogén de inmunoglobulina epsilon que fue reportado (Ueda et al, PNAS 82: 3712 1985) como compartido por el gorila y el ser humano, pero no por el chimpancé, pareciendo contradecir la visión evolutiva convencional de que los antepasados humanos divergieron de la línea del gorila antes de divergir de la línea del chimpancé. Desafortunadamente, Woodmorappe no consideró datos posteriores del laboratorio de Ueda (Kawamura y Ueda, Genomics 13:194, 1992) que estaban disponibles cuando Woodmorappe escribió en 1994 (Bible Science News 32:4 p. 12). Estos datos más recientes muestran que las deleciones de ADN que destruyeron copias duplicadas de los genes de inmunoglobulina epsilon (1) ocurrieron independientemente en las líneas humanas y de gorila (la independencia se dedujo del hecho de que los límites "derecho" e "izquierdo" del ADN delecionado eran completamente diferentes en las dos especies), y (2) también ocurrieron (otra vez independientemente) en el chimpancé. Por lo tanto, el ejemplo de Woodmorappe de un pseudogén compartido que vincula a los humanos con el gorila pero no con el chimp (en aparente violación de la divergencia más reciente de los antepasados humanos del chimpancé aceptada por la mayoría de los evolucionistas) es incorrecto: estos no son pseudogenes "compartidos" sino pseudogenes que surgieron independientemente, y el chimpancé tiene una deleción similar, aunque más grande. (Debo mencionar que cité este mismo ejemplo incorrecto en mi versión original de este ensayo. Sin embargo, en el momento en que lo escribí--1986--el ejemplo estaba respaldado por la evidencia disponible en ese entonces; e imprimí una corrección en Creation/Evolution después de que se publicaran los nuevos datos. También debo enfatizar que el ejemplo del pseudogén de epsilon procesado mencionado en la sección 4.3 anterior representa una secuencia completamente diferente, que nadie disputa que es compartida por humanos, chimpansés y gorilas.) |
Sin embargo, el ejemplo de pseudogenes compartidos que Woodmorappe ofrece para desafiar el modelo evolutivo tiene una explicación más mundana: se basa simplemente en información incorrecta y desactualizada (véase cuadro 3).
5.9 Una hipótesis final ofrecida por el Sr. Woodmorappe (en correspondencia personal) es que genomas similares (como los del humano y el chimpancé) podrían tender a adquirir los mismos pseudogenes de forma independiente, mientras que genomas menos similares podrían ser menos capaces de adquirir los mismos pseudogenes. Esta hipótesis, obviamente ad hoc, teóricamente explicaría por qué—even si los humanos y los chimpancés fueron creados independientemente—podrían compartir más pseudogenes que pares de especies menos similares y relacionadas independientemente, como el humano y el gibón. El problema con esta hipótesis es que la ocurrencia independiente—es decir, en dos individuos diferentes—de la misma retroposona insertándose en la misma posición casi nunca ha sido reportada, incluso en individuos de la misma especie. He podido encontrar solo cuatro publicaciones que describen ejemplos de inserciones idénticas e independientes. Una involucra un virus de sarcoma de Rous modificado, diseñado con un marcador seleccionable específico e infectando fibroblastos de pavo cultivados en cultivo de tejidos (Shih et al, Cell 53:531, 1988); e incluso en este papel inusual con un virus especialmente diseñado, la frecuencia de tales inserciones se estimó en solo 1 de cada 4000 eventos de inserción. El segundo ejemplo es una publicación muy reciente y controvertida (Slattery et al., Mol Biol Evol 17:825, 2000) que interpreta dos inserciones idénticas de un SINE en la misma ubicación (un intrón del gen Smcy) en un gato doméstico y un gato montés como representando inserciones independientes en lugar de reflejar una única inserción en un felino ancestral común. Dos publicaciones adicionales (Kass et al, J Mol Evol 51:256 2000; Cantrell et al., Genetics 158:769, 2001) describen aparentes inserciones idénticas pero independientes de SINEs en especies de ratón. (John Woodmorappe se negó a citar ningún dato cuando fue desafiado a proporcionar ejemplos de inserciones independientes. Sin embargo, si los lectores de este ensayo son conscientes de otra evidencia sobre inserciones independientes del mismo elemento en la misma posición en cualquier modelo de laboratorio, agradecería las citas apropiadas y actualizaré este ensayo para reflejarlas.) Muy muchas inserciones que ocurren naturalmente han sido documentadas en elementos TY de levadura, gypsy y elementos P de drosophila, retrovirus y transgenes murinos, e inserciones de VIH humano—todas sin haber sido reportadas inserciones independientes idénticas. Si organismos independientes de la misma especie (es decir, con genomas más idénticos que el humano versus el chimpancé) casi nunca adquieren el mismo pseudogeno o inserción de retroposona en la misma posición, es difícil tomar en serio la hipótesis de que, por ejemplo, las mismas siete inserciones Alu en las mismas posiciones del locus globín humano y chimpancé ![]() (ver sección 4.4 arriba) podrían haber ocurrido como 14 eventos de inserción independientes.
(ver sección 4.4 arriba) podrían haber ocurrido como 14 eventos de inserción independientes.
5.10 ¿Podría un pseudogén haber sido transmitido por un virus de una especie a otra, dando lugar a pseudogenes compartidos? Una propuesta a lo largo de estas líneas ha sido sugerida por el anti-evolucionista Pat Kohli y parece superficialmente plausible. Varios virus, incluidos los retrovirus, se sabe que ocasionalmente captan secuencias de nucleótidos de una célula "donante", las cuales luego, tras la reinfección de una nueva célula, pueden insertarse en el ADN de la nueva célula "receptora". De hecho, este mecanismo se sabe que tiene consecuencias significativas: si el ADN transmitido incluye una versión mutada de ciertos genes clave que regulan la división celular, tal secuencia de ADN puede actuar como un oncogén y causar malignidad en la célula receptora (Bishop, Cell 42:23, 1985). Teóricamente, una secuencia de pseudogén o retroposón podría ser capturada por un virus y luego transmitida entre especies mediante este mecanismo, dando lugar a la existencia de secuencias inútiles idénticas compartidas entre dos especies. De hecho, se han reportado casos raros de transferencia aparente entre especies de retroposones (por ejemplo, entre dos especies de moscas de la fruta [Jordan et al, PNAS 96: 12621, 1999] o de una serpiente venenosa a los rumiantes [Kordis y Gubensek, PNAS 95:10704, 1998; Kordis, Genetica 107:121, 1999]). Sin embargo, esto no es probablemente la explicación para la mayoría de los pseudogenes/retroposones compartidos por al menos tres razones.
Primero, los pseudogenes/retrotransposones compartidos se encuentran generalmente en la posición exactamente homóloga en el ADN de cada especie. Esto es casi siempre cierto en el caso de los pseudogenes clásicos, que se encuentran en estrecha proximidad al gen funcional, y también es cierto para las secuencias Alu como las mencionadas en el clúster del gen globina, y para los pseudogenes procesados cuya ubicación ha sido determinada (por ejemplo, el pseudogen procesado de inmunoglobulina epsilon humano mencionado anteriormente [Ueda, et al, EMBO J 1:1539, 1982; Tanabe et al. Cytogenet Cell Genet 73:92, 1996]). Los sitios diana para la inserción viral pueden compartir ciertas características de secuencia local (Craigie en Trends in Genetics 8:187, 1992; Knoblauch et al., J Virol 70:3788, 1996; Stevens y Griffith, PNAS 91:5557, 1994), pero estas características ocurren con bastante frecuencia y generalmente están dispersas por todo el ADN de la célula receptora. Aparte de los artículos mencionados en la sección 5.9, no hay precedente ni mecanismo conocido para que un segmento de ADN transmitido por un virus dirija un sitio específico en el ADN de la célula receptora, como sería necesario para que un pseudogen que representa una inserción viral hipotética ocurra en la misma ubicación que la secuencia donante hipotética. Por lo tanto, las ubicaciones compartidas de pseudogenes/retrotransposones con respecto al ADN circundante argumentan fuertemente en contra de tal modelo de transmisión entre especies.
En segundo lugar, si la mayoría de los pseudogenes/retrotransposones compartidos representaran transferencia mediada por virus de una especie a otra, se esperaría encontrar secuencias virales cerca de pseudogenes en especies "receptoras". Dichas secuencias virales están regularmente presentes en ejemplos conocidos de transmisión viral de ADN de una célula a otra, incluyendo inserciones de construcciones retrovirales diseñadas; pero no se encuentran secuencias virales asociadas con la mayoría de los pseudogenes/retrotransposones, con excepción de los retrovirus endógenos.
Finalmente, se han generado varios árboles genealógicos comparando entre especies la presencia o ausencia de inserciones LINE o Alu en ubicaciones específicas del genoma, un ejercicio similar al mostrado en la Figura 5 anterior (Malik et al, Mol Biol Evol 16: 793, 1999; Hamdi et al J Mol Biol 284:861, 1999); los árboles genealógicos derivados de retrotransposones fueron exactamente congruentes con los árboles previamente establecidos basados en similitudes de secuencias y características anatómicas. Si la transferencia entre especies explicara la mayoría de los retrotransposones compartidos, no se esperaría tal congruencia.
En una búsqueda asistida por computadora de la literatura científica, solo pude encontrar dos ejemplos de pseudogenes para los cuales la transmisión viral fue incluso tentativamente considerada como un mecanismo de origen, en ambos casos con evidencia bastante débil (Gruskin et al., PNAS 84:1605, 1987 y Robins et al., J Biol Chem 261:18, 1986). Los lectores que estén conscientes de otros ejemplos son invitados a enviarlos por correo electrónico para su inclusión en futuras actualizaciones de este artículo. Por el momento, la evidencia argumenta en contra de la transferencia interespecífica mediada por virus como un mecanismo general para explicar los pseudogenes/retrotransposones compartidos.
5.11 El creacionista L. J. Gibson también ha abordado los pseudogenes en un artículo publicado (Origins 21:91, 1994). El artículo de Gibson se reduce a dos puntos, uno similar al discutido en la sección 5.2 anterior, más un punto adicional de carácter más filosófico. Él señala que el argumento de los pseudogenes compartidos se basa en la suposición de que "Dios no crearía secuencias no funcionales similares en especies separadas", lo cual él llama "un argumento teológico [que] apenas puede ser abordado por la ciencia" y que requeriría apoyo bíblico para ser creído. Dado que la Biblia no aborda —y por lo tanto deja abierta— la posibilidad de que Dios pueda crear secuencias no funcionales en el ADN, Gibson siente que no se puede descartar la noción de que Dios de hecho creó tales secuencias individualmente a medida que creó cada especie, incluidas aquellas secuencias no funcionales que ahora encontramos compartidas entre diferentes especies.
Debe mencionarse parenthéticamente que el argumento de Gibson socava la propia interpretación de los creacionistas sobre la similitud de las especies mencionada al principio de este ensayo (sección 1.2). Como discutimos, los creacionistas han afirmado que los árboles de similitud basados en información de secuencia no deben aceptarse como evidencia de relaciones evolutivas, porque las especies creadas independientemente por un diseñador inteligente podrían esperarse que muestren patrones idénticos de aparente parentesco. La crítica de Gibson se aplica igualmente contra este argumento creacionista, ya que la Biblia no menciona los planes de Dios sobre la similitud de secuencias.
Sin embargo, como discutimos anteriormente, esta noción creacionista de secuencias similares diseñadas para funciones similares tiene al menos algún sentido intuitivo. En contraste, Gibson propone una hipótesis ad hoc claramente inaceptable cuando sugiere que un diseñador podría haber colocado inserciones de retrotransposones no funcionales --imitando todas las características de aquellos que actualmente se retrotransponen aleatoriamente en el laboratorio-- en las mismas posiciones del ADN de especies creadas independientemente; esta idea merece tanta credibilidad como la afirmación de que las entradas falsas compartidas en un directorio se deben a errores independientes en lugar de plagio. La hipótesis de Gibson no argumenta a favor de un creador que toma decisiones de diseño comprensibles, sino de un creador tan impredecible que podría ser el autor de cualquier hallazgo científico tradicionalmente interpretado como no diseñado --a menos que la Biblia lo especifique de manera diferente. Por lo tanto, la lógica de Gibson respondría a las siguientes afirmaciones, ya que no son específicamente contradichas por la Biblia: (1) Dios creó fósiles que parecen los restos de animales que nunca vivieron, y los incrustó en rocas. (2) Dios creó elementos radiactivos en rocas que sugerirían falsamente edades mayores que sus edades reales. (3) Dios creó el universo hace 6000 años con la luz de las estrellas en camino a nuestros ojos pero con las propiedades esperadas de la luz que salió de estrellas hace miles de millones de años. En otras palabras, la lógica de Gibson nos invita a rechazar cualquier argumento científico a favor de la evolución si ese argumento no está específicamente verificado en la Biblia. La visión de Gibson puede ser internamente consistente, pero claramente requiere que la verdad de la Biblia sea aceptada por fe como base para juzgar los méritos de las conclusiones científicas, y por lo tanto se aparta de la verdadera ciencia basada en la prueba de hipótesis, la lógica inductiva y conclusiones basadas en datos observados. Si un científico ve una inserción de retrotransposón en el laboratorio como resultado de varios parámetros bioquímicos conocidos, la navaja de Occam lo desalienta de postular una mano inteligente guiando su creación. Si encontramos otras inserciones en nuestro ADN con características idénticas a aquellas que surgen bajo observación, asumimos que las que están en nuestro ADN surgieron por un mecanismo similar. Sabemos que tales inserciones que surgen bajo observación en el laboratorio pueden usarse para rastrear la linaje de animales de laboratorio, y que otras inserciones naturales pueden usarse para rastrear poblaciones en la naturaleza; no tenemos razón para disuadirnos de usar inserciones similares para rastrear las linajes de diferentes especies. Este razonamiento inductivo es fundamental para la paleontología, la datación radiométrica, la astronomía, la física, la medicina y cada otro campo de la ciencia. Si Gibson siente que los méritos de un argumento científico dependen de cuán bien sea apoyado por la Biblia, puede simplemente descartar la evolución en su totalidad porque entra en conflicto con el Génesis, y evitar la molestia de tratar con todos los detalles de las observaciones y deducciones científicas individuales en las que se basa la evolución. Este enfoque de "la Biblia primero" puede ser apropiado para la religión, pero es inaceptable como ciencia.
6. Poniendo a prueba el modelo
Una característica de la ciencia que la distingue de la creencia religiosa revelada (y de los evolucionistas de los creacionistas) es la convicción científica de que se puede obtener nuevo conocimiento sobre el pasado mediante un análisis cuidadosamente diseñado del mundo moderno. Los creacionistas a menudo afirman que, dado que el origen de las especies ocurrió en un pasado remoto, no existe una manera científicamente válida de estudiar el proceso hoy en día y, por lo tanto, la evolución no es una ciencia real que pueda ser sometida a prueba mediante experimentos. Sin embargo, incluso sin experimentos reales, una hipótesis científica puede ser sometida a prueba si sugiere una predicción no trivial que pueda verificarse o falsarse mediante la recopilación de más datos.
De hecho, la interpretación de los pseudogenes procesados compartidos descrita aquí representa una hipótesis que puede ser probada porque presenta una implicación bastante sorprendente: al comparar dos secuencias de nucleótidos de una sola especie—that is, las secuencias de un pseudogen procesado y el gen funcional del cual se originó—debería ser posible predecir qué otras especies compartirán el mismo pseudogen y cuáles no. Para comprender la lógica de tal predicción, considere el hecho de que si un pseudogen procesado surgió en una especie antigua, se deberían encontrar copias de ese pseudogen en los descendientes modernos de dicha especie. Por lo tanto, según el modelo evolutivo, si supiéramos cuándo surgió un pseudogen procesado humano y pudiéramos así fijar su origen en una posición particular en el árbol evolutivo aceptado, predeciríamos que el mismo pseudogen procesado debería encontrarse en las especies modernas que derivan de ese punto del árbol y no en ninguna otra rama.
De hecho, existe una forma de estimar cuándo se formó un pseudogén procesado dado. Resulta que las mutaciones "silenciosas"—es decir, mutaciones que no tienen efecto sobre la supervivencia del organismo (como todas las mutaciones en pseudogenes inútiles)—se acumulan a una tasa bastante uniforme. Esta tasa ha sido estimada examinando el número de diferencias de secuencia "silenciosas" entre secuencias funcionales correspondientes en dos especies y comparando este número con la fecha aproximada de divergencia de las mismas dos especies según lo indicado por el registro fósil. Dada esta tasa de mutación y el número de diferencias de secuencia entre un pseudogén procesado particular y su gen fuente funcional (del mismo especie), se puede estimar la fecha de origen del pseudogén; luego, basándose en esta fecha, se pueden derivar predicciones sobre qué otras especies modernas deberían portar el mismo pseudogén. Estas predicciones pueden probarse buscando el pseudogén en una variedad de especies.
Consider, for example, the processed human epsilon pseudogene discussed earlier (section 4.3). The number of nucleotide differences between this pseudogene and the functional gene suggests that this pseudogene arose about 40 million years ago. Therefore, the evolutionary interpretation of processed pseudogenes presented in this essay would predict that mice and rabbits (which are thought to have diverged from the human lineage 70 to 80 million years ago, before the apparent origin of the pseudogene) should not carry this pseudogene. In contrast, apes and Old World monkeys--whose estimated dates of divergence from the human lineage (5-10 and 30 million years ago, respectively) are both after the apparent pseudogene origin--would be expected to carry the pseudogene. Available evidence confirms all of these predictions and is also consistent with similar predictions about the species distribution of other processed pseudogenes (see for example Anagnou et al. PNAS 81:5170, 1984 con respecto a la dihidrofolato reductasa, Craig et al. Gene 99:217, 1991 con respecto a la isomerasa del trifosfato de triosa, y Friedberg y Rhoads Molec. Phylogenet & Evolution 16:127, 2000 con respecto a la enolasa, la calmodulina y la sintetasa de argininosuccinato).
Por una lógica similar, es posible estimar la edad de inserción de un retrovirus endógeno comparando las secuencias de los LTR "izquierdo" y "derecho" (véase la sección 2.2.2.d anterior). Dado que el LTR "izquierdo" se copia a partir de la secuencia del LTR "derecho" en el momento de la inserción, ambos LTR comparten una secuencia idéntica en el momento en que se origina la copia del retrovirus. Tras la inserción, los dos LTR acumulan mutaciones de forma independiente, por lo que el número de diferencias de secuencia entre los dos LTR puede utilizarse para estimar la edad de una inserción retroviral particular; esta edad puede utilizarse para predecir la distribución de especies de copias compartidas de la inserción retroviral particular. Cuando recientemente se estimaron las edades de varios retrovirus endógenos humanos utilizando este enfoque, se confirmó la distribución de especies predicha de las copias compartidas (Johnson & Coffin, PNAS 96:10254, 1999).
Aunque las inserciones individuales de SINE y LINE en humanos no pueden fecharse fácilmente mediante el análisis de secuencias por sí solo, puede ser posible estimar un período de tiempo aproximado cuando ciertas subclases se insertaron. Esto es posible porque se cree que clases específicas de retrotransposones similares poblaron el genoma de los mamíferos en oleadas, con ciertas familias (y subfamilias, en el caso de las secuencias Alu y LINE) siendo copiadas de un pequeño número de retrotransposones fuente activos en cualquier período particular de nuestra historia evolutiva. Este modelo se ha deducido del hecho de que para algunas familias de retrotransposones humanos (por ejemplo, LINE2), las comparaciones entre miembros específicos de la familia revelan secuencias relativamente divergentes, como si las copias individuales hubieran acumulado muchas mutaciones diferentes durante mucho tiempo desde su inserción en nuestro ADN (Nature 409:860, 2001, ver pág. 881), mientras que otros, como las secuencias Alu (particularmente las subfamilias Ya5 y Ya8) y la familia Ta de secuencias LINE1, muestran menos desviaciones de una secuencia consenso y, por lo tanto, se cree que se insertaron más recientemente. Estas interpretaciones basadas en la evolución de las secuencias de retrotransposones humanos predicen un amplio compartir de especies de retrotransposones presumiblemente más antiguos, pero un compartir más restringido de los presumiblemente más jóvenes, y estas predicciones se confirman con datos independientes de distribución de especies para estos retrotransposones (Gonzalez et al, Genomics 18:29, 1993; Shaikh y Deininger, J Mol Evol 42:15, 1996; Carroll et al, J Mol Bio 311:17, 2001; Sheen et al. Genome Res 10:1496, 2000; Boissinot et al., Mol Biol Evol 17:915, 2000).
Se descubrirán con certeza más retroposones compartidos, y solo el tiempo dirá cuán consistentemente se confirman predicciones evolutivas como estas. Pero por ahora, casi todos los datos disponibles son consistentes con los modelos evolutivos de descendencia común, y no se ha propuesto ninguna alternativa creacionista para explicar esta consistencia. Los casos repetidos de este tipo de predicción y confirmación pueden proporcionar evidencia convincente para la evolución, incluso si algunos tipos de experimentos directos, como estudios sobre dinosaurios vivos, son imposibles. (Si los lectores son conscientes de otros ejemplos de pseudogenes procesados u otros retroposones cuya distribución en diferentes especies apoya o contradice el "árbol genealógico" aceptado, agradecería escuchar sobre estos casos por correo electrónico, y revisaré esta publicación según corresponda.)
7. Conclusión
¿Demuestran las secuencias compartidas y sin función descritas aquí que los humanos y los simios tuvieron un ancestro común? En realidad, ningún conocimiento científico se basa en una prueba irrefutable del tipo que respalda los teoremas matemáticos, por lo que la queja creacionista de que la evolución "nunca ha sido probada" simplemente revela un malentendido grave sobre la naturaleza de la ciencia. Más bien, la ciencia avanza mediante la acumulación de pistas buscadas por detectives persistentes (científicos) que intentan deducciones lógicas e imparciales a partir de estas pistas. Como un jurado presentado con estas pistas, podemos intentar llegar al veredicto más probable, aunque reconozcamos que nuestros hechos son incompletos; no hay "testigos" vivos de los eones de evolución, por lo que debemos hacer las mejores deducciones posibles a partir de las pistas disponibles. En "el caso de las secuencias compartidas sin función", un jurado imparcial concluiría seguramente que copiar de un ancestro compartido era la explicación más probable, consistente con la interpretación evolutiva. Esta conclusión seguiría la lógica de la ley de derechos de autor real, en la que los errores compartidos se aceptan como evidencia de copia. La fuerte aceptación de esta conclusión entre los científicos está indicada por el hecho de que no se ha propuesto ninguna explicación alternativa en la literatura científica para explicar la amplia compartición de tantas secuencias sin función entre especies. Por lo tanto, si debemos aceptar la evidencia de la ciencia, parece que la descendencia común de especies dispares de un ancestro compartido ("macroevolución" en la terminología creacionista) ha ocurrido realmente.
A medida que los genetistas moleculares descubren nuevos ejemplos de pseudogenes compartidos y retrotransposones, esta información se unirá al inmenso conjunto de pistas provenientes de otras disciplinas que, en conjunto, ya proporcionan una evidencia abrumadora a favor de la evolución. A pesar de esta impresionante evidencia, ningún científico cree que todas las respuestas sobre la evolución estén ya disponibles o que nuestro conocimiento actual sobre los pseudogenes y los retrotransposones esté inmune a revisiones a la luz de futuros conocimientos. De hecho, los científicos en laboratorios de todo el mundo continúan explorando los genes de diversas especies, comparando los datos de la genética molecular con el registro fósil y refinando nuestro conocimiento de la historia de nuestra especie.
En la etapa actual de esta investigación interminable, la evidencia sugiere lo que para mí es una noción asombrosa: como una piedra de Rosetta biológica o un rollo del Mar Muerto, nuestro propio ADN —un valor equivalente a una Enciclopedia Británica de información en cada célula de nuestro cuerpo— contiene un registro del pasado que apenas estamos aprendiendo a leer. Este registro, que refleja millones de años de historia genética, incluye los restos de antiguos accidentes genéticos que ocurrieron antes de que nuestros antepasados similares a los monos recorrieran las sabanas de África, restos con los que ahora compartimos otros descendientes de esos mismos antepasados: los gorilas modernos y los chimpancés.
| Las preguntas frecuentes | Archivos que deben leerse | Índice | Creacionismo | Evolución | Edad de la Tierra | Geología del diluvio | Catastrofismo | Debates |